




Introduction
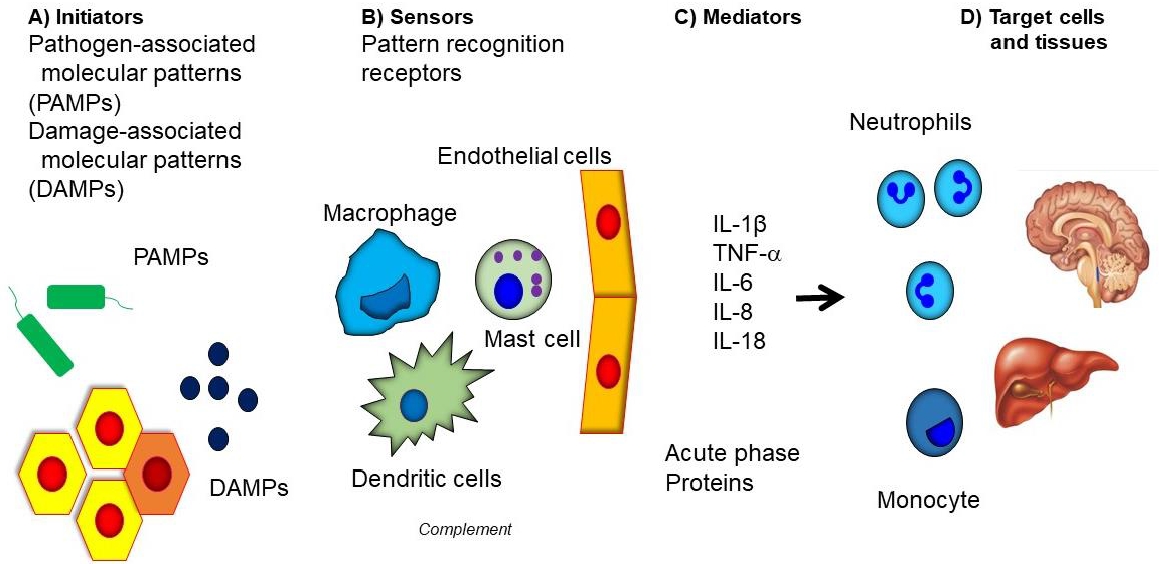
Inflammation is a defense mechanism that protects against harmful external factors such as pathogens or injury [1,2]. Inflammation has been documented from time immemorial and is accompanied by symptoms such as redness, swelling, heat and pain [2]. It comprises four stages of innate immunity: inflammatory inducers, the sensors for inducers, inflammatory mediators, and target tissue that are affected by mediators (Fig. 1). After an inflammation process, the inducer is removed, and the function of the sensor during the inflammatory process disappears through negative feedback. In the absence of inflammatory mediators, the inflammatory response stops at the target tissue. When there is a genetic mutation in the inflammatory process, the acute inflammation does not disappear, and repeated acute inflammation induce tissue or organ damage [3,4]. In chronic inflammation, the inflammatory response is continuously maintained through various environmental factors or etiologies, resulting in damage to surrounding organs [5].
Systemic autoinflammatory disorder (SAID) is a group of disorders caused by a dysregulation of the innate immune system during the inflammation process [4,6-8]. The disorders are induced by abnormal activation of innate immunity without infection or autoimmunity [3,6]. SAID represents persistent and recurrent acute inflammation that occurs through an antigen-independent pathway. One of best recognized SAID is Familial Mediterranean fever (FMF), which was discovered in 1945. The term “autoinflammatory” was first used in 1999 [9]. Recently, SAID has been identified as a single gene mutation, and many cases are accompanied by autoimmune or allergic manifestations and immunodeficiency [4,6,10,11].
Molecular mechanism of inflammation and autoinflammation
SAID develops due to overactivation of the innate immune system. Inflammation in innate immune cells occurs through the pattern recognition receptors (PRRs) on cells for pathogen associated molecular pattern by pathogen, and damage associated molecular pattern that causes tissue damage. PRR has membrane bound Toll-like receptors and C-type lectin receptors, and its intracellular sensors include nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), absence in melanoma 2-like receptors, and pyrin, which constitute multimeric protein scaffolds called inflammasomes. An inflammasome is composed of apoptosis-related speck-like proteins containing caspase activation and recruitment domains (ASC), which are adaptor molecules, and use caspase-1 as effector molecules [4,10,12,13].
The NLR consists of a NOD domain, and a leucine-rich repeat domain, and a caspase recruitment domain (CARD), which exists in the C-terminal. Since ASC comprises the pyrin domain and CARD, caspase-1 is recruited and activated through CARD. Activated caspase-1 is released into the blood stream, converting pro-interleukin (IL)-β to activated IL-1β. When IL-1β binds to the IL-1 receptor, active nuclear factor-κB (NF-kB) is moved to cell’s nucleus by Myeloid differentiation primary response 88, and as a result, the transcription of various genes that induce an inflammatory response occurs, leading to an inflammatory response [13,14].
The interferon (IFN) system can also lead to inflammation due to innate immunity to viral or bacterial infections. Type I IFN is induced by microbial or viral nucleic acid, and the nucleic acid is sensed in the cytoplasm or endosome by double-strand (ds) DNA or ds RNA. After this, IFN regulatory factors are activated and translocated into the nucleus, and this finally induces the transcription of IFN. Type I IFNs bind to the IFN receptor, and activate signal translation by Janus kinase (JAK)/signal transducers and activators of transcription pathway. The IFN plays an essential function in noncanonical NLRP3 (nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain 3) inflammasome activation and pyroptosis [4,11,14].
Clinical manifestations of SAIDs
Patients with SAID present with either recurrent or periodic flares of systemic inflammation with fever and dramatic elevation of acute phase reactants. Further, various skin rashes, serositis, and lymphadenopathy may develop with variable durations of symptom free intervals [4,6,15]. They can be triggered by stress, sleep deprivation, and antigenic stimulation after vaccination or viral infections. Some patients present with more chronic manifestations. Dermatologic findings help with the diagnosis as various dermatologic features show pseudo-urticarial rash, vasculitis rash, and granulomatous dermatitis [7,16]. Recently, the clinical features of monogenic SAID combined with immunodeficiencies with atuoinflammation [11,17].
SAID includes monogenic, and polygenic disorders such as PFAPA (periodic fever, aphthous stomatitis, pharyngitis, adenitis) syndrome, systemic juvenile idiopathic arthritis, adult-onset Still disease, and Behcet disease [6]. Recently, adult-onset SAID were reported as Schnitzler syndrome [18], and vacuoles, E1 enzyme, X-linked, autoinflammatory and somatic (VEXAS) syndrome [19].
Diagnostic approach
Most patients with SAID present with recurrent or periodic fever with inflammatory manifestations for long periods. Conditions such as malignancies, infections, autoimmunity, immune dysregulation, and drug hypersensitivity must be excluded. Further, details regarding the fever are needed, such as its onset, frequency, regularity, duration, severity, and triggering factors including vaccination or stress. After that, growth and development must be considered because recurrent infections can induce growth impairment [4,6]. Family history, including consanguinity family deaths, and immunologic diseases is very important for genetic study [20]. While assessing history, accompanying characteristics and their timing must be described including skin rash, fever pattern, and gastrointestinal or musculoskeletal manifestations such as arthritis [6].
Disease categories according to their mechanism
1. IL-1 and inflammasomopathy
IL-1 and inflammasomopathy result from predominant innate immune system dysregulation through a monogenic problem. Patients with inflammasomopathy present with “periodic or recurrent fever syndrome” with inflammatory symptoms or signs and skin manifestations [3,12,14]. The skin manifestations are mostly urticarial-like rash, characterized by neutrophil infiltration around vessels without eosinophils (Table 1) [16,21].
2. Endoplasmic reticulum stress
The clinical manifestations differ according to genetic mutations and function, although the pathogenesis depend on endoplasmic reticulum function defect. The TRAPS (tumor necrosis receptor-associated periodic syndrome) causes recurrent fever, abdominal pain, arthralgia, and other inflammasomopathy-like signs and symptoms [22]. The COPA (coatomer associated protein subunit alpha) syndrome causes pulmonary hemorrhage or interstitial lung disease, autoimmune manifestations, and renal dysfunction (Table 2) [23].
3. Mutations in endogenous antagonists
This results from the absence of down-regulation of IL-1 or IL-36 due to a dysfunction or the absence of receptors. This defect of IL-1- or IL-36-related inhibition is associated with early-onset hyperinflammation, and severe skin manifestations such as pustular skin lesions and vasculitis (Table 3) [24,25].
4. Dysregulation of NF-κB signaling
The NF-κB activation in immune response is controlled by signaling and ubiquitination/deubiquitination balance. An uncontrolled NF-κB pathway induces hyperresponsiveness to cytokines, and leads to autoinflammation [26,27]. Human RelA haploinsufficiency induces increased apoptosis in response to tumor necrosis factor (TNF) and impaired NK-κB activation [28]. The unbalanced ubiquitination/deubiquitination system due to mutation leads to an overactivation of NF-κB by a defective negative feedback pathway [27]. Most cases with dysregulation of NF-κB dysregulation signaling present with early-onset autoinflammatory manifestations such as fever, and arthritis (Table 4) [27,29].
5. Type I interferonopathy
The clinical manifestations of type I interferonopathy develop early with autoimmune findings, including necrotizing vasculitis, noninfectious interstitial lung disease, panniculitis with or without lipodystrophy, and thrombotic events [30]. The clinical findings differ from inflammasomopathy, such as lymphopenia, skin manifestations, and vasculopathy [30-32]. Aicardi-Goutieres syndrome, as typical type I interferonpathy in neonatal period, is characterized by basal ganglia calcification without congenital infections [30]. The level of auto-antibodies here is relatively lower than that in adult-onset autoimmune diseases, and involved in early-onset kidney or lung disease (Table 5) [30-32].
6. Adult-onset SAIDs
1) Schnitzler syndrome [18]
This syndrome is the acquired autoinflammatory syndrome without germline NLRP3 mutations. The clinical features include chronic urticarial rash, joint or bone pain, lymphadenopathy, and fever with immunoglobulin M monoclonal gammopathy. And these symptoms are similar to those of cryopyrin-associated periodic syndrome.
2) VEXAS syndrome [19]
This syndrome is a monogenic adulthood autoinflammatory disease with hematologic manifestations. The pathogenesis is related to somatic mutations in UBA1, which encodes for the master enzyme of cellular ubiquitylation. The VEXAS syndrome comprises macrocytic anemia, thrombocytopenia, and thromboembolic events with progressive bone marrow failure.
Genetic approach to SAID diagnosis
SAID is related to strong genetic backgrounds, and are controlled by the innate immune system [3,12]. Molecular confirmation has now become essential for their clinical diagnoses [4,6]. The clinical manifestations in most SAID are modulated according to age by environmental and epigenetic influences, although SAID is associated with genetic mutations [33]. Genetic studies regarding SAID are based on the following: (1) clinical criteria for known monogenic SAID, (2) partial presentation with family history of SAID, (3) suspicion of SAID with irreversible damage, and (4) strong suspicion of SAID even though there is no fulfilled clinical criteria [20,34].
Approach to therapy
Most SAID can be controlled by steroids, but steroid usage is limited in pediatrics because of growth defects and other serious adverse effects. The treatment for SAID depends on the control of over-expressed immune reactants based on its primary pathogenesis [35,36]. Colchicine can be used to control the inflammation that results from SAID. The mode of action of colchicine is thought to result from impairing microtubules and the expression of adhesion molecules, thus reducing neutrophil trafficking and down regulating the inflammatory pathway [37]. The recommended dose in European Alliance of Associations for Rheumatology is as follows: starting dose of <0.5 mg/day for children <5 years, 0.5–1.0 mg/day for children 5–10 years, 1.0–1.5 mg/day in children over 10 years and adults. The patients are monitored every 6 months for their complete blood cell counts, and renal and liver functions [38].
IL-1 blockade is effective in disorders for inflammasomopathies such as cryopyrin-associated periodic syndrome, and deficiency of the interleukin-1 receptor antagonist, among others [39]. Anakinra is a short-acting IL-1β blocking agent, and canakinumab is long-acting type [39,40]. Patients are monitored every month for first 3 months and then every 3 months for complete blood cell counts, and renal and liver functions [6,39].
TNF blockade may be effective for TNF-related SAID. TNF blockade can only be done after the exclusion of latent tuberculosis. Further, monitoring is done every 3-6 months for complete blood cell counts and comprehensive metabolic panel [6,35,36].
The JAK inhibitor for mediating IFN signaling may also be useful for interferonopathy. JAK inhibitors have been shown to reduce signs of panniculitis, and lipodystrophy, and have induced remission in some cases. When these inhibitors are used, the presence of BK or herpes virus infections, and the liver and renal functions have to be monitored [30,32,35].
Conclusion
The diagnosis of SAID is based on various signs and symptoms according to immune dysfunction. However, the clinical manifestations of SAID include allergies, autoimmunity, and immunodeficiency. Molecular diagnosis in SAID should be considered early in evaluating patients with any suspected monogenic cause of autoinflammation, and expert advice is required for appropriate test selection and interpretation. Personalized therapeutics in SAID can be achieved through molecular diagnosis, optimization of medication dosing, and guidance based on the evidence of grading of available literature in addition to expert opinion.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


