

Introduction
Nonketotic hyperglycinemia (NKH) is an autosomal recessive inherited inborn error of amino acid metabolism characterized by elevated concentrations of glycine in plasma, urine, brain, and cerebrospinal fluid (CSF) due to a defect in the glycine cleavage enzyme system, a four-protein complex responsible for the interconversion of the amino acids glycine and serine. The activity of this enzyme system is deficient in the livers and brains of affected patients1,2).
Children with NKH develop severe neurological symptoms. After a short period of good health, muscular hypotonia, lethargy, poor feeding with hiccups, and intractable seizures ensue. Progression to respiratory insufficiency, apnea, coma, and death usually follows. Most patients experience a fulminant disease course, and lethal symptoms often develop during early postnatal life. Children who survive have severe developmental abnormalities, mental retardation, refractory seizures, and hypotonia that can progress to spasticity1-3). Pharmacologic treatment remains unsatisfactory, even when initiated early. Prospective treatment can modify the early neonatal course of severe NKH but does not prevent the devastating neurological outcomes, suggesting glycine-induced prenatal injury and ongoing postnatal damage4,5).
The characteristic mark of NKH is an increased glycine concentration in the plasma and CSF, leading to an elevation of the CSF: plasma glycine ratio>0.08 (normal, <0.04). A confirmed diagnosis can be made using an enzyme assay of liver tissue or by mutational analysis; however, this is invasive and occasionally impractical6,7).
Magnetic resonance spectroscopy (MRS) provides a noninvasive biochemical presentation of tissue by showing normal and pathological metabolite patterns. MRS is a new tool to examine metabolic diseases converting compounds in the visible spectrum8,9). This report demonstrates that MRS can be used to measure glycine directly and noninvasively in the brain tissue of patients suffering from NKH and may be useful for monitoring treatment.
Case report
A male baby was delivered at 40 weeks gestation by emergency caesarean section due to progression failure. The Apgar scores at 1 and 5 minutes were 6 and 8, respectively. The pregnancy had been uncomplicated, and the infant was morphologically normal, with a birth weight of 3,230 g (50th percentile), length of 52 cm (75th percentile,) and head circumference of 35 cm (75th percentile). The parents were not consanguineous, and had no family history of metabolic or neurologic diseases. A paucity of movement, weak crying, and intermittent hiccups were observed in the first 24 hours. At that time, the patient had generalized hypotonia and twitching movements. On day 3, he became lethargic and began shallow respirations that progressed to frequent apnea. A blood-gas analysis, standard biochemistry tests, and inflammatory markers were normal. Brain ultrasonography revealed partial agenesis of the corpus callosum, megacisterna magna, and abnormal echogenecity in the brain. On day 4 of life, an amino acid analysis of the plasma was requested to exclude the possibility of a metabolic disorder. On day 7, the amino acid analysis revealed glycine levels of 1,710 ┬Ąmol/L (normal, 232 to 740 ┬Ąmol/L). The patient was transferred to our neonatal intensive care unit for further evaluation and treatment on day 7.
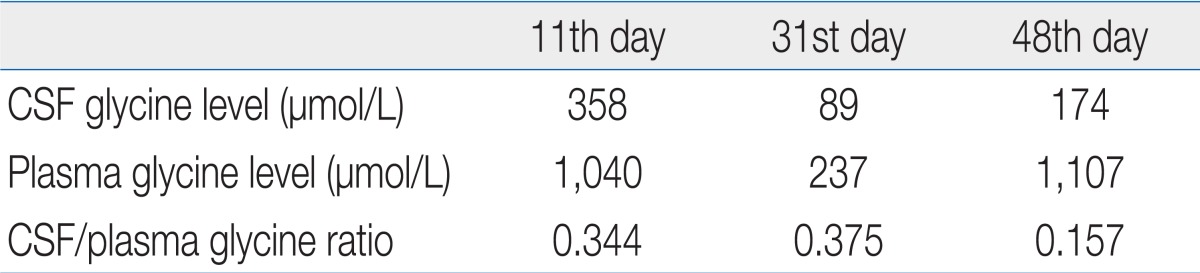
On admission, he was lethargic, hypotonic, and apneic. Stimulus-sensitive myoclonus, twitching movements involving all four limbs, and recurrent hiccups were observed. Plasma amino acid profiles obtained on days 7 and 11 revealed elevated glycine levels of 2,408 and 1,040 ┬Ąmol/L (normal, 232 to 740 ┬Ąmol/L), respectively. His CSF amino acid profile was 358 ┬Ąmol/L (normal, <13 ┬Ąmol/L) on day 11 and the CSF/plasma glycine concentration ratio was 0.34 (normal, <0.04), which was diagnostic for NKH.
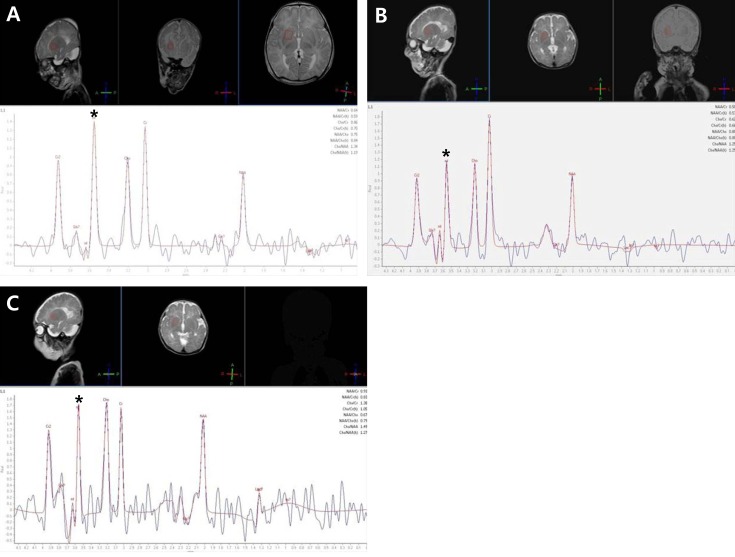
On day 11, brain magnetic resonance imaging (MRI) and MRS were performed using a 3.0-tesla system. The MRI scan showed high-signal intensity in the bilateral perirolandic white matter and internal capsule on T1-weighted and fluid attenuated recovery images, suggesting abnormal myelination. Thinning of the corpus callosum and mega cisterna magna were also observed. MRS and long echo time spectra were obtained at echo times of 135 ms. At short echo times, a consistent time-domain analysis failed due to the strong signal overlap of glycine with myo-inositol resonance. The spectra were compared with those from an age-matched control subject. The patient's MR spectrum produced markedly increased peak intensity at 3.55 ppm, which was assigned to a glycine peak, consolidating the diagnosis of NKH. We used the glycine/creatinine area ratio (control value, 0.60)10,11), as determined by serial MRS, to estimate cerebral glycine levels. Glycine/creatinine area ratios were obtained at the frontal white matter, basal ganglia, and parietal white matter and were 0.912, 1.135, and 1.802, respectively (Figs. 1, 2).
On day 11, an endotracheal intubation was performed due to frequent apnea. An exchange transfusion with 1 unit of blood was performed to rapidly decrease serum glycine levels. Subsequently, the patient was treated with sodium benzoate (250 mg/kg/day), dextromethorphan (7.5 mg/kg/day), parenteral amino acid-free nutrition, and a glycine-free formula. His symptoms improved after intensive treatment. Respiration improved slowly over several days and the patient was extubated on day 20. On day 31, a follow-up amino acid analysis revealed decreased CSF and plasma glycine concentrations of 89 and 237 ┬Ąmol/L, respectively (Table 1, Fig. 2). A follow-up MRS showed a decreased amount of metabolite (glycine) in the basal ganglia and parietal white matter but increased metabolites in the frontal white matter. The glycine/creatinine area ratios were 0.645 in the basal ganglia, 1.249 in the parietal white matter, and 1.729 in the frontal white matter (Figs. 1, 2).
On day 36, the sodium benzoate was tapered and stopped on day 43. The patient again developed frequent seizures and apnea. On day 48, an amino acid analysis showed increased CSF and plasma glycine concentrations up to 174 and 1,107 ┬Ąmol/L, respectively (Table 1, Fig. 2). On day 48, a follow-up MRS showed an increased glycine/creatinine area ratio of 1.377 in the basal ganglia but decreased ratios in the frontal white matter and parietal white matter of 1.133 and 0.611 respectively (Figs. 1, 2). MRI on day 48 showed persistent abnormal signal intensity in the white matter and moderate cerebral atrophy. The patient was treated with sodium benzoate again and symptoms improved.
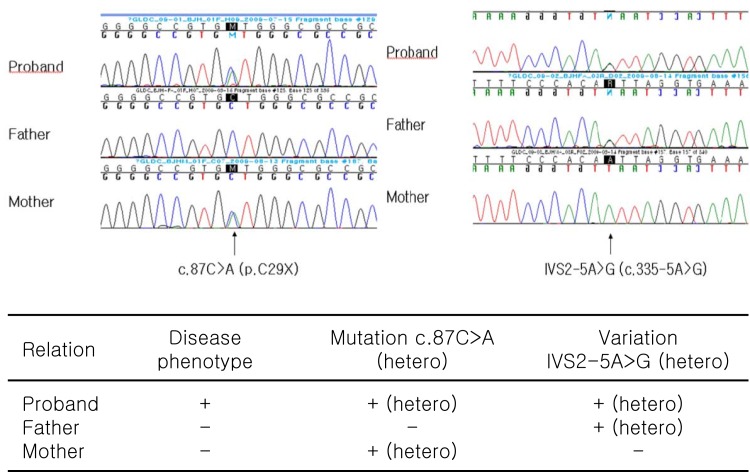
A genetic analysis was performed by direct sequencing of all exons and introns of the glycine decarboxylase (GLDC) gene using DNA extracted from peripheral blood leukocytes. As a result, two variations within the GLDC gene were found, one of which was thought to be a novel missense mutation (c.87C>A). Another variation (IVS2-5A>G) was identified in the intron within the GLDC gene, but no conclusive data supported it as a disease-causing mutation (Fig. 3).
He was discharged home on hospital day 70. A follow-up at 28 months revealed disastrous neurological outcomes. His weight and length were within normal limits, but head circumference was <3rd percentile. He did not attain any milestones expected for his age and was dependent on a gastric tube for feeding. Intractable seizures with erratic myoclonus and tonic spasms became nearly continuous. He has been treated with multiple antiepileptic drugs including phenobarbital, valproic acid, oxcarbazepine, vigabatrin, clobazam, and a ketogenic diet but not much improvement was observed.
Discussion
NKH is caused by an inherited deficiency of the glycine cleavage system and is characterized by the accumulation of a large amount of glycine in the body, particularly in the CSF. High concentrations of glycine within the central nervous system distinguish NKH from other forms of hyperglycinemia. In patients with NKH, glycine accumulates in the brain and spinal cord at very high levels, which is reflected in the CSF. In other forms of hyperglycinemia, brain and CSF glycine levels remain normal despite high levels of plasma glycine. Because children with NKH usually do not have neurological symptoms, the high concentrations of glycine within the CNS have been considered responsible for the neurological symptoms3,12).
Glycine serves as a inhibitory neurotransmitter in the CNS13) and is also a coagonist of glutamatergic neurotransmission at the N-methyl-D-aspartate (NMDA) glutamate receptor subtype. High levels of glycine in the brain overstimulate the NMDA receptor system, and the excitatory attributes of glycine play an important role in glutamate-induced neurotoxicity. Glycine stimulates recovery of the receptor following glutamate-induced desensitization and, therefore, increases the frequency of NMDA receptor channel opening. Therefore, glycine multiplies the excitotoxic activity of glutamate, which is supposed to be a result of an excessive elevation of intracellular free Ca1,14,15). Excessive activation of excitatory amino acid receptors, particularly the NMDA receptor, is involved in the pathogenesis of neuronal injury and destruction in numerous neurologic disorders including hypoxia-ischemia, hypoglycemia, and brain trauma. In particular, the developing brain shows increased susceptibility to NMDA-mediated brain injury, and high levels of glycine may be particularly devastating to the CNS of a neonate16,17).
The diagnosis of NKH is established by evaluating glycine levels in plasma and CSF. CSF glycine can be more than 30 times that of normal in patients with the neonatal form of NKH. Plasma glycine in untreated patients can be very high but occasionally is within the normal range. The elevated ratio of CSF to plasma glycine distinguishes NKH from other glycine metabolism disorders6). However, as there is a relatively higher glycine level in plasma compared with CSF, an elevated CSF glycine level in the presence of CSF blood is not interpretable12). Additionally, brain injury, hypoxia-ischemia, severe malnutrition, the use of anticonvulsant medications, and other inborn errors of metabolism (including ketotic hyperglycinemia) can also result in elevated levels of CSF glycine and may confuse the diagnosis of NKH. The confirmative test for diagnosis is an open liver biopsy, demonstrating deficient glycine cleavage system activity; however, this procedure is not practical in many cases6,12). A genetic analysis can also be used to confirm the diagnosis; however, it requires a substantial amount of time7).
Reports on image findings related to NKH have been insufficient because of the scarcity and poor prognosis of this disease18). Press reported that MRI studies of seven patients with NKH revealed progressive atrophy and delayed myelination. No correlation has been found between the degree of volume loss or abnormality of myelination demonstrated by MRI and the glycine concentration in the CSF or plasma. Routine brain MRI findings are usually normal during the acute phase of this disease, despite serious clinical symptoms19).
MRS has been widely used in various kinds of metabolic brain diseases to examine specific abnormal metabolites. However, in the majority of such diseases, the results have been non-specific, particularly from a diagnostic point of view. NKH is one of the few metabolic brain diseases in which MRS can detect a specific metabolite that accumulates in the brain. In fact, MRS is a unique technique that can noninvasively quantify glycine in the brain in vivo. Furthermore, MRS directly demonstrates abnormally high levels of glycine in the brain parenchyma in NKH, being reflected as an abnormal peak at 3.55 ppm8,9).
Many studies have reported that glycine levels measured by MRS correspond to changes in plasma glycine levels and to clinical features. Heindel et al.8) reported that glycine in brain tissue corresponds more reliably with clinical findings than stable plasma and CSF values. He found that glycine concentrations in plasma and CSF after the first month of life are relatively constant, whereas MRS from the basal ganglia and parieto-occipital white matter change in accordance with clinical findings. Suzuki et al.20) examined a girl with NKH using MRS and revealed a time course of glycine/creatinine ratios similar to the changes in glycine levels in the blood and CSF. They suggested that MRS is a noninvasive diagnostic tool useful for directly monitoring brain glycine levels in patients with NKH. Snider et al.9) obtained serial MR spectra of parietal white matter from patients with NKH and also found good correlations between glycine/choline and glycine/creatinine ratios estimated by MRS and clinical and biochemical course.
MRS spectra are usually obtained in the basal ganglia or parietal white matter in most research involving MRS for NKH. It is difficult to compare glycine concentrations using MRS in different regions of the brain. Previous reports demonstrated that the measured glycine concentration varies within different parts of the brain, with higher levels in the spinal cord, cerebellar cortex, and basal ganglia compared to those in the cerebral cortex3,21). Huisman compared quantitative MR spectra obtained from different brain regions and chemical quantifications of brain tissue obtained by autopsy. The quantitative chemical analysis of different brain regions corresponded with the MRS results, and the glycine concentration in basal ganglia was the highest21).
In our case, the patient had three times the serial CSF and serum glycine levels, and MRI and MRS were obtained at the same time. We initially observed that glycine concentration increased, peaking at 3.55 ppm, which was sufficient to confirm the diagnosis of NKH. Furthermore, MRS enabled a quantitative measurement of glycine levels in different parts of the brain, and we found that the value taken from the basal ganglia closely reflected the CSF and plasma glycine concentrations (Fig. 2).
In conclusion, MRS allowed for the non-invasive measurement of glycine concentrations in the brain at multiple sites. Thus, MRS can distinguish NKH from other metabolic disorders exhibiting increased plasma glycine levels. Additionally, MRS is a useful tool to monitor the effects of therapeutic interventions.

 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


