


Introduction
X-linked adrenoleukodystrophy (X-ALD) is the most common peroxisomal disorder, it is a rapidly progressive, neurodegenerative, and recessive disease that characteristically primarily affects the central nervous system white matter and the adrenal cortex1,2). The prevalence of this condition is generally reported to be between 1:100,000 and 1:20,000, although its overall frequency may be as high as 1:17,0003,4). There are three major phenotypic forms of the disease, which is rapidly progressive childhood cerebral form, a milder adrenomyeloneuropathy in adults, and Addison disease only5). These clinical features are manifestation of impaired peroxismal β-oxidation, which results in increased levels of saturated unbranched very long chain fatty acid (VLCFA) in the plasma and tissue, and particularly in the cholesterol ester, ganglioside, and proteolipid fractions of the brain white matter and in the cholesterol ester fraction of the adrenal cortex2).
X-ALD is diagnosed with clinical, radiological, and serological tests, including tests for elevated plasma VLCFA such as C24:0 and C26:0, and high C24:0/C22:0 and C26:0/C22:0 ratios1,2,3,4,5), which are complemented with genetic analyses. The X-ALD gene, ABCD1, has 10 exons and is located on chromosome Xq28, and was identified with a positional cloning strategy1). To date, more than 600 ABCD1 mutations have been identified.
We report a 7.5-year-old Korean boy with the childhood cerebral form of X-ALD and an unpublished mutation in the ABCD1 gene, despite normal plasma concentrations of VLCFA.
Case report
A 7.5-year-old boy was admitted to to Department of Pediatrics, Chungnam National University Hospital with weakness of the bilateral lower extremities. The patient was a healthy child, born at term. He appeared to be healthy for his first six years, and his developmental milestones were normal. His family history included no neurological disease. Two months before admission, he occasionally fell over his foot and showed leg weakness. Five days before admission, he could not stand or walk by himself. Neurological examinations revealed that grade was four per five of motor-power at upper extremities and three per five at lower extremities. He showed hyperreflexia of bilateral knee and ankle jerks. The patient spoke inarticulately. No skin hyperpigmentation were apparent.
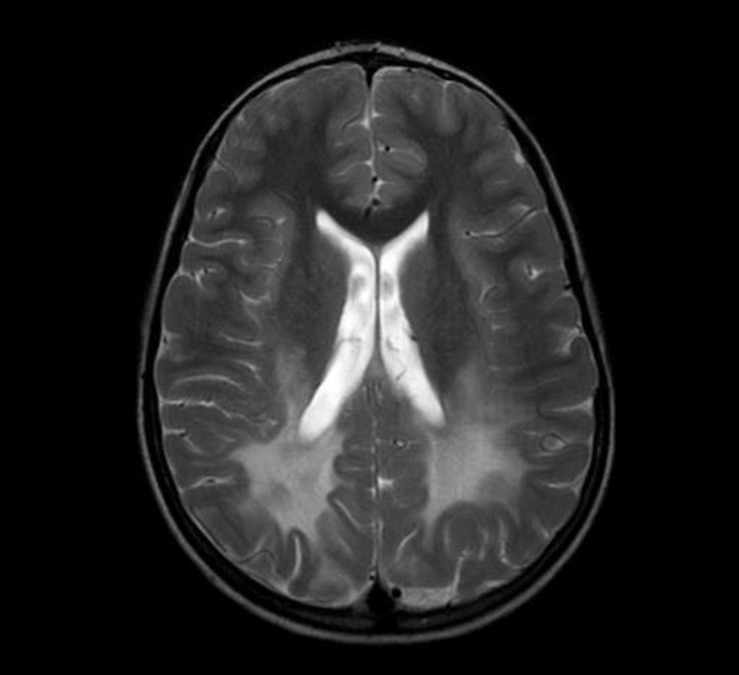
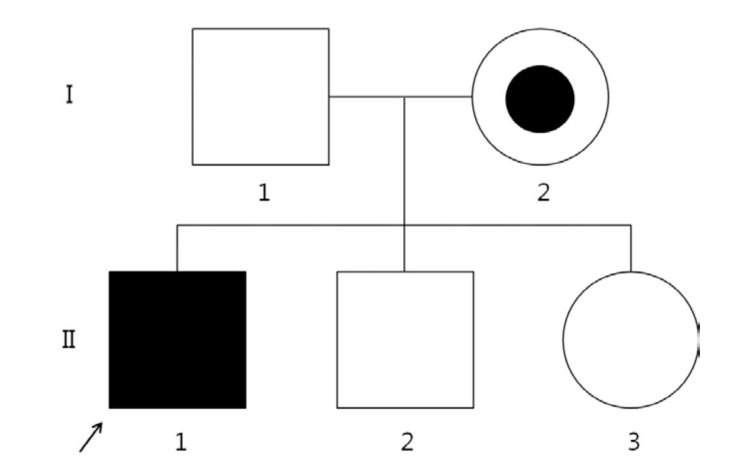
Brain magnetic resonance imaging revealed a confluent T2-weighted high-signal-intensity lesion in the parieto-occipitofronto-temporal periventricular white matter, especially, around the atrium of the lateral ventricle, the posterior half of the corpus callosum, and posterior limb of internal capsule, and along the pyramidal tract of the brainstem (Fig. 1). The proband was clinically suspected of suffering ALD. Laboratory tests revealed normal morning serum cortisol (8.72 µg/dL; normal, 5-25 µg/dL) and elevated adrenocorticotropic hormone (126.3 pg/mL; normal, 9-52 pg/mL). His serum glucose, Na+, and K+ levels were within the normal ranges. The 1 µg-synacthen test showed a peak cortisol response of 17.02 µg/dL, confirming primary partial adrenal insufficiency. The fasting plasma levels of VLCFA, measured with gas chromatography-mass spectrometry, were: C22:0, 11.954 µmol/L (normal, 0-96.300 µmol/L); C24:0, 19.234 µmol/L (normal, 0-91.400 µmol/L); and C26:0, 0.893 µmol/L (normal, 0-1.310 µmol/L). His C24:0/C22:0 and C26:0/C22:0 ratios were significantly elevated at 1.609 (normal, 0-1.390) and 0.075 (normal, 0-0.023), respectively. Genemic DNA was extracted from peripheral whole blood samples collected from the patient and his family. All samples were taken after informed consent was obtained. The splice junctions and all coding regions of the ABCD1 gene of the proband were amplified by polymerase chain reaction (PCR) using specific primers, but only exon 1 was amplified and sequenced in family members I-1, I-2, II-2, and II-3 (Fig. 2). The PCR products were directly sequenced using the same primer pairs and the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA), according to the manufacturer's instructions. DNA sequences were analyzed using an ABI 3130×l Genetic Analyzer (Applied Biosystems). The ABCD1 gene in the proband revealed a missense mutation (p.Arg163Leu) caused by the nucleotide change 488G>T in exon 1 (Fig. 3A). His asymptomatic mother carried the same mutation (Fig. 3B). His younger brother and sister had no mutation in the ABCD1 gene. The c.488G>T (p.Arg163Leu) substitution is a previously unpublished mutation in the ABCD1 gene.
He treated with Lorenzo's oil (3-4 spoon/day, 1 vial/mo; Scientific Hospital Supplies Co., San Juan, Trinidad and Tobago), hydrocortisone, lovastatin, and Coenzyme-Q. The benefic and adverse effect of Lorenzo oil and lovastatin were not shown. After 2 years, he is in wheel-chair based doing, not bed-ridden. And he treats with rehabilitation for four limb rigidity and spasticity, swallowing difficulty, and decreased cognition.
Discussion
Although this patient showed imaging findings of typical ALD, his biochemical findings revealed VLCFA levels within normal limit. His diagnosis was based on his clinical features and increased C26:0/C22:0 and C24:0/C24:0 ratios. This diagnosis was confirmed by genetic evidence of an ABCD1 mutation.
This mutation in the ABCD1 gene, c.488G>T (p.Arg163Leu) in exon 1, has been reported by Dr. Steinberg through the internet-accessible mutation database for X-ALD (http://www.x-ald.nl). However, He has not published this datum. This is the first published report of the c.488G>T mutation in exon 1 of the ABCD1 gene.
Since the establishment of the X-ALD Database, 1236 mutations have been reported in the ABCD1 gene, 582 (47%) of which appear to be cryptic, but these mutations are not distributed evenly throughout the entire gene6). They included 51% missense mutations, 11% nonsense mutations, 28% frameshift mutations, 6% amino acid insertions/deletions, and 3% deletion of one or more exons7). There is no clear correlation between these mutations and the phenotypes of ALD patients6,7).
The main biochemical abnormality and reliable diagnostic tool is a high plasma concentration of VLCFA in exclusively male X-ALD patients. Plasma VLCFA can be evaluated with three parameters: the concentration of C26:0, the ratio of C26:0 to C22:0, and the ratio of C24:0 to C22:0. All three VLCFA parameters are elevated in 99% of male with X-ALD8). The database website for X-ALD (http://www.x-ald.nl) demonstrated that approximately 10% to 20% of obligate carriers (women with X-ALD) have normal levels of VLCFA. However, a normal plasma VLCFA level thus does not exclude heterozygosity for X-ALD. In our patient, his plasma C26:0, C24:0, and C22:0 levels were all in the normal ranges, and only his C26:0/C22:0 and C24:0/C22:0 ratios were elevated. Asian reports with ABCD1 gene mutation in exon1 revealed that all males with X-ALD had increased levels of VLCFA9). Interestingly, only our case showed normal range of VLCFA levels. Because it is difficult to assess the correlation between phenotype and genotype, the role and meaning between missense mutation and normal levels of VLCFA has not solved in our case. Kennedy et al.10) reported that the sensitivity of measures of plasma VLCFA levels is less than 100% for X-ALD. They also categorized X-ALD into three subgroups, according to the plasma VLCFA levels: "typical ALD", "normal", and "in need of further study". In the borderline results, that define the subgroup, "in need of further study", any one of the three parameters has a normal value. Our patient falls into the "in need of further study" subgroup of X-ALD, and we designed this disease "atypical X-ALD". The concentrations of plasma VLCFA levels do not also correlated with the phenotypes of the ALD patients8,10).
In conclusion, if the clinical suspicion of X-ALD is strong plasma VLCFA levels are highly diagnostic measure. However, if the results for VLCFA are consistent with atypical ALD, mutational analysis of the ABCD1 gene can confirm the proper diagnosis of X-ALD. We have reported a Korean boy with atypical X-ALD, confirmed by a previously unpublished datum from the X-ALD Database, c.488G>T (p.Arg163Leu) in exon 1 of ABCD1.

 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


