


Introduction
Parathyroid hormone (PTH) is the principal physiological regulator of calcium. It maintains the plasma calcium level by reabsorbing calcium from the bones and kidney and promoting the production of active vitamin D1). Insufficient production of PTH can induce hypocalcemia. The etiology of hypoparathyroidism is diverse. In comparison with adults, in whom damage after thyroid surgery is a common cause of hypoparathyroidism, a congenital disorder is the most common cause in childhood2). DiGeorge syndrome, which is associated with deletion of chromosome 22q12, mutations in the glial cells missing homologue B (GCMB) gene, HDR syndrome (hypoparathyroidism, sensorineural deafness, renal dysplasia) caused by mutation of the GATA3 gene, HRD syndrome (hypoparathyroidism, retardation, dysmorphism) caused by a defect in the TBCE gene, familial X-linked recessive hypoparathyroidism, mutations of PTH genes, and Kearns-Sayre syndrome resulting from mutations in mitochondrial DNA are known causes of congenital hypoparathyroidism1,2). Mutations in the calcium sensing receptor (CaSR) can also cause hypoparathyroidism1,2,3).
CaSR is a 1,078 amino acid G-protein-coupled receptor which consists of an extracellular domain of 612 amino acids, seven transmembrane domains of 250 amino acids and an intracellular domain of 216 amino acids. Although calcium is the endogenous ligand of CaSR, other cations such as Mg2+, Ba2+, and Gd3+ can also bind CaSR4). The action of CaSR bound to ions other than Ca2+ is unknown3). CaSR is widely distributed, but the highest levels are found in the parathyroid gland and kidney tubular cells. Its main function is to maintain calcium homeostasis5). In the parathyroid gland, parathyroid cells detect small perturbations of the extracellular calcium concentration and modulate the secretion of PTH in an inhibitory manner. Extracellular calcium concentration and PTH secretion have an inverse sigmoidal relationship with each other5). In the kidney, CaSR is highly expressed on the basolateral side of the cortical thick ascending limb of the loop of Henle (TALH) in rats6). A high peritubular calcium concentration activates CaSR and activated CaSR inhibits transporters in the TALH, which subsequently reduces Ca2+ and Mg2+ reabsorption7). Inactivating and activating mutations of CaSR can occur. Inactivating mutations lead to resistance to extracellular calcium, which causes hypercalcemic disorders including familial hypocalciuric hypercalcemia or neonatal severe hyperparathyroidism8). Activating mutations in the CaSR lead to hyperresponsiveness to extracellular calcium, which causes hypocalcemia with hypercalciuria, a disorder called autosomal dominant hypocalcemia (ADH)9). Activating mutations of CaSR may also cause a Bartter phenotype by inhibition of transporters in the TALH. The association of ADH with Bartter syndrome (BS) has been previously described10,11). The present case report describes a patient with ADH and type V BS due to an activating mutation in the transmembrane domain of CaSR.
Case report
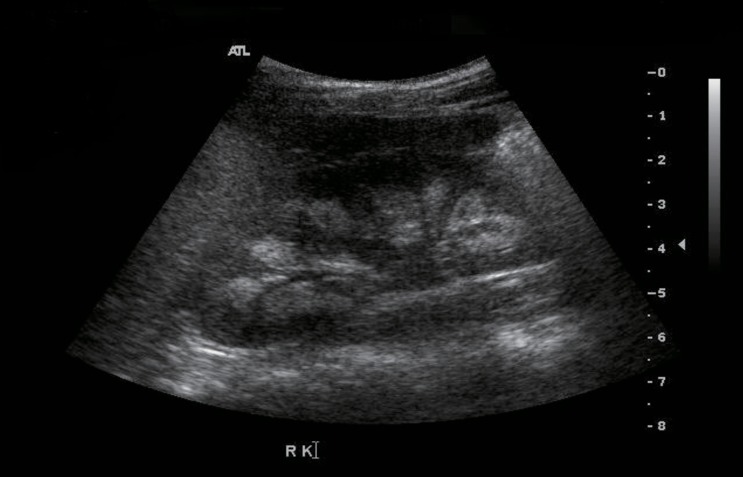
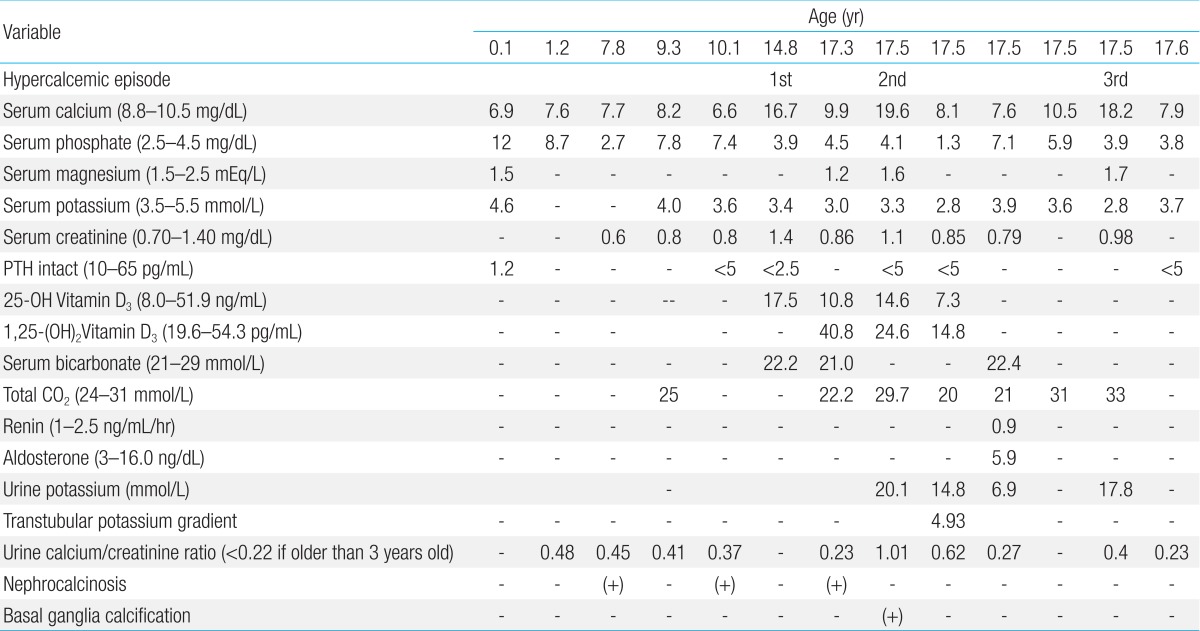
The patient was followed at the Seoul National University Children's Hospital, Inje University Busan Paik Hospital and Maryknoll Medical Center for hypocalcemia, hypokalemia, and hypomagnesemia. The disorder was detected soon after birth. She had generalized seizures on the twelfth day after birth. She attended Dong-A University Hospital and laboratory results showed hypocalcemia, hypomagnesemia, and a low level of PTH. Oral calcium, magnesium, and vitamin D supplements were prescribed and the seizures were controlled. However, if she stopped her medication, she had a seizure. Thiazide was added because of hypercalciuria and medullary nephrocalcinosis (Fig. 1). At 14 years of age, she suffered weight loss from 48 kg to 43 kg and a loss of appetite. After three days of nausea and vomiting, she attended hospital. Laboratory testing showed a serum calcium level of 16.9 mg/dL and serum phosphorus of 3.9 mg/dL. Her serum creatinine was 1.4 mg/dL and her serum blood urea nitrogen 22.8 mg/dL (Table 1). She had been prescribed calcium carbonate 1,000 mg/day and alfacalcidol 1.5 µg/day before admission (Table 2). Medication was discontinued until serum calcium and phosphorus levels were below the normal range, after which the medications were restarted.
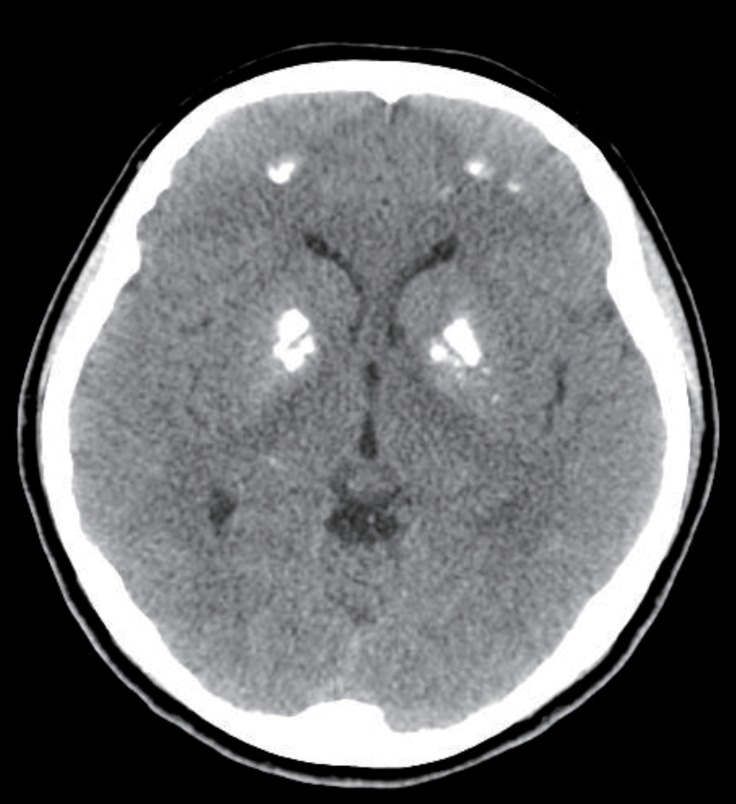
At 17 years old, the patient visited hospital complaining of dizziness, nausea, and vomiting. Her serum calcium level was 19.6 mg/dL and her serum potassium 2.8 mg/dL. Calcification of the subcortical and basal ganglia of both frontal lobes was found on brain computed tomography (Fig. 2, Table 1). Her medication at admission was calcium acetate (4,000 mg/day), alfacalcidol (2.5 µg/day), hydrochlorothiazide (75 mg/day), and amiloride (6.6 mg/day) (Table 2). She had moved from Maryknoll Medical Center to Inje University Busan Paik Hospital in Busan a month before the second hypercalcemic episode and the dose of alfacalcidol had been increased from 1.25 µg/day to 2.5 µg/day at her initial visit to Inje University Busan Paik Hospital because her serum 25-hydroxy-vitamin D3 (25-OHVitD3) concentration was just above the lower limit of the normal range. All medication was discontinued and her serum calcium and potassium concentrations gradually normalized, but because of persistent dizziness she was transferred to Seoul National University Children's Hospital. On the day after transfer, she complained of paresthesia of her hand and her serum calcium concentration was 8.2 mg/dL. Alfacalcidol and calcium carbonate were administered and the dose of medication was slowly increased to 2 µg/day of alfacalcidol and 3,000 mg/day of calcium carbonate. Alfacalcidol was changed to calcitriol 1.0 µg/day and she was discharged with 1.0 µg/day of calcitriol, 3,000 mg/day of calcium carbonate, and 50 mg/day of thiazide. In the outpatient clinic 2 days after discharge, her calcium carbonate was reduced to 2,000 mg/day because her serum calcium level was elevated to 10.5 mg/dL.
Ten days after her outpatient clinic visit, she was admitted again because of high calcium level. She did not complain of muscle weakness, nausea, or vomiting and she did not appear anxious or depressive. Her height was 153.4 cm (5th-10th percentile) and weight 50 kg (25th-50th percentile). She was being treated with oral calcium carbonate (2,000 mg/day), calcitriol (1.0 µg/day), and hydrochlorothiazide (50 mg/day) (Table 2). Her serum calcium concentration was 18.2 mg/dL (normal range, 8.8-10.5 mg/dL), serum phosphorus 3.9 mg/dL (normal range, 2.5-4.5 mg/dL), serum magnesium 1.7 mEq/L (normal range, 1.2-5 mEq/L), and serum potassium was 2.8 mmol/L (normal range, 3.5-5.5 mmol/L). Her PTH level was below 5 pg/mL (normal range, 10-65 pg/mL) (Table 1). All medications were withheld and intravenous hydration was started. Medications were restarted when her calcium concentration reached 9.2 mg/dL and the calcium concentration was maintained at a stable level with 2,000 mg/day of calcium carbonate, 1.5 µg/day of alfacalcidol, 12.5 mg/day of hydrochlorothiazide, and 1,800 mg of potassium chloride.
After informed consent was obtained, direct sequencing of all coding exons of CASR revealed that the patient had a heterozygous substitution of adenine (TAT) for guanine (TGT) at codon 829, which would result in substitution of cysteine for tyrosine (Y829C) in the protein (Fig. 3).
Her parents and brother did not have hypocalcemia on laboratory testing. Her mother underwent genetic study and did not have a mutation in CaSR.
Discussion
This patient had ADH with BS because of a Y829C activating mutation of the CASR gene. This mutation has not been reported previously.
Activating mutations in the CaSR can cause BS10,11). BS is a genetically heterogeneous disorder with a unifying pathophysiology of decreased salt absorption by the TALH. About 30% of sodium and chloride filtered in the glomeruli is reabsorbed in the TALH. Defects in the function of proteins associated with sodium reabsorption in TALH cause BS12). However, reabsorption not only of sodium, but also of water and of other ions, including calcium, magnesium, and potassium, is influenced. Five genes are known to be associated with BS10,12). Type 1 BS is caused by a loss-of-function mutation of the apical sodium-potassium-chloride cotransporter gene (NKCC2). Type 2 BS is caused by mutations in the apical renal outer medullary potassium channel gene (ROMK). Mutations in the basolateral chloride channel (CLCNKB) gene cause type 3 BS. Type IV BS results from a loss-of-function mutation of BSND, which encodes barttin, the β subunit of CLC-K channels that is essential for their function. Type V BS is caused by activating mutations of CASR10,12). CaSR expressed in the TALH is activated by a high extracellular concentration of calcium ions, and activated CaSR inhibits the NKCC2 and ROMK in the apical membrane and the sodium potassium ATPase in the basolateral membrane of the TALH12). In rat kidney, cytochrome P-450-dependent metabolites of arachidonic acid and cyclic AMP are associated with the inhibitory effect of CaSR on ROMK13). Calcium and magnesium in the TALH are reabsorbed by a paracellular pathway driven by a positive luminal potential. Potassium excretion through ROMK is essential to generate this positive luminal voltage relative to the basolateral side, so inhibition of ROMK blocks the generation of the higher voltage on the luminal side and increases urinary calcium and magnesium excretion10,12).
Several mutations have been identified in the CASR gene10,11,14,15). The clinical presentations and onset timing of Bartter's phenotype differ according to the type of mutation10). Patients with K47N or P221L mutations demonstrate hypocalcemia without clinical features of BS. Another case reported a patient with K29E mutation, who presented with hypocalcemia and mild hypokalemia without alkalosis, hyperreninemia, and hyperaldosteronemia. In this patient, hypokalemia occurred at 22 years old and was corrected with small doses of oral potassium11). Patients with A843E or C131W mutations show the phenotype of BS including nephrocalcinosis, hypokalemia, hypomagnesemia, metabolic alkalosis, hyperreninemia, and hyperaldosteronemia as well as hypocalcemia. In patients with A843E mutation, the features of BS develop before 6 years old. Each type of mutated CaSR has different functional characteristics10). The extracellular calcium concentration at which CaSR shows half of its maximal activity (EC50) was 2.0-2.2 mmol/L (8.0-8.8 mg/dL) for CaSR with K47N or P221L mutations, 1.45 mmol/L (5.8 mg/dL) with the K29E mutation11), and below 0.5 mmol/L (2.0 mg/dL) for CaSR with A843E or C131W mutations. The EC50 of the wild-type CaSR is 3.4 mmol/L (13.6 mg/dL). The lower the EC50, the higher is the activity of CaSR. This explains why patients with A843E or C131W mutations have the most severe symptoms of BS. The onset timing of phenotype of BS also correlates with EC50; the lower the EC50, the faster the onset of Bartter's phenotype10,11).
The characteristics of BS were expressed mildly in this patient. Serum magnesium concentrations were in the subnormal to low-normal range. Serum potassium concentrations maintained in the normal range until hypokalemia appeared when the patient was about 17 years old. Since then, the patient has been taking oral potassium. Metabolic alkalosis or hyperaldosteronism did not occur. This phenotype is similar to that of CaSR with the K29E mutation, so it could be assumed that the EC50 of CaSR with the Y829C mutation is similar to that of CaSR with the K29E mutation. However, this should be cautiously interpreted, as BS can differ between patients who have the same CaSR mutation11). The patient in this case had nephrocalcinosis and basal ganglia calcification. In one study of patients with CaSR mutation, basal ganglia calcification was found in 9 of 25 patients (36%) and nephrocalcinosis was present in 3 of 25 patients (12%)14). In another study, nephrocalcinosis occurred in 8 of 14 patients (57%). Hypercalciuria was associated with nephrocalcinosis15), but basal ganglia calcification and nephrocalcinosis were not related to the severity of hypocalcemia14).
This patient had three hypercalcemic episodes. The first episode occurred when she was 14 years old and the others when she was 17 years old. Hypercalcemic episodes during the treatment of ADH have been reported previously14,15), and occurred in patients who had taken a high dose of alfacalcidol. During her first hypercalcemic episode, our patient suffered fever and severe weight loss. The volume loss and the continuous medication might have caused the hypercalcemia. Before the second event, her dose of alfacalcidol was increased because of low serum 25-OHVitD3, although her serum calcium concentration remained in the normal range. Thus, an overdose of alfacalcidol might have induced the hypercalcemia. In the third hypercalcemic episode, the patient denied overdose of medication, infection, or over consumption of foods high in calcium. The cause of hypercalcemia in this third event is not clear. But, considering the increase of serum calcium concentration from 7.6 mg/dL to 10.5 mg/dL 2 days after discharge, the doses of calcium and vitamin D3 supplementation were probably too high for the patient.
Fine adjustments are needed in the treatment of ADH. Symptomatic hypocalcemia should be treated to raise serum calcium concentration. When symptoms disappear, medication doses should be adjusted to maintain serum calcium above the level that symptoms persisted. When increasing the doses of medications, special attention should be taken not to cause hypercalcemia.
In conclusion, our paper reports a novel Y829C mutation of CASR. The patient with this CASR gene mutation had a mild phenotype of BS in addition to ADH.


 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


