


Introduction
Estimates for asphyxia-related neonatal deaths vary from 0.7 to 1.2 million annually1), and peripartum asphyxia is a major cause of disabilities and sensorineural impairments2,3,4). Hypoxic-ischemic encephalopathy (HIE) is an important source of permanent damage to central nervous system tissues that may result in neonatal death or manifest later as cerebral palsy or developmental delay5). In fact, about 20%-30% of infants with HIE die in the neonatal period, and 33%-50% of survivors are left with permanent neurodevelopmental abnormalities such as cerebral palsy and mental retardation5).
There are a number of in vivo and in vitro studies on the protective effects of ischemic preconditioning in which a period of sublethal hypoxia followed by another ischemic event 1 to 2 days later results in protection from an otherwise lethal insult6,7,8,9,10,11,12). Gidday et al.10) first showed that exposure of 8% oxygen to neonatal rat pups for 3 hours had a protective effect on a stroke produced by carotid occlusion and oxygen. According to Khaspekov et al.11), an episode of 60 minutes of oxygen-glucose deprivation (OGD), followed 1 and 2 days later by 90 minutes of OGD, resulted in 40%-60% protection in mixed neuroglial hippocampal cell cultures.
These previous reports have demonstrated that certain percentages of oxygen, exposure times to sublethal hypoxia, and the extent of sublethal hypoxia varies across studies. The hippocampus is comprised of a simple anatomic structure that is easy to manipulate. As a result, organotypic hippocampal slice cultures are commonly used to study neuronal cell death, neuroprotection, and synaptic plasticity13). Organotypic slice cultures have several advantages, which include the ability to preserve the gross cytoarchitecture of the hippocampus as well as the possibility of long-term observation and manipulation, which are typical requirements examining physiological or pathological processes14).
To investigate the effects of and to designate the extent of mild hypoxia, we exposed rat organotypic hippocampal slice cultures in immature and mature brains to 10% O2 for 60 minutes.
Materials and methods
1. Organotypic slice culture
Using the Stoppini method15), organotypic slice cultures of the hippocampus were prepared from a total of 12 Sprague Dawley rats (DooYeol Biotech, Seoul, Korea) aged 7days. Rats were stabilized for 24 hours before the start of the experiment and were decapitated using scissors. Heads were sterilized with 70% alcohol, and the hippocampi were quickly removed and placed in ice-cold Gey's Balanced Salt Solution (GBSS, Sigma Aldrich, St Louis, MO, USA). The hippocampi were sliced (450 ┬Ąm thick) using a tissue chopper (Stoelting Co., Wood Dale, IL, USA), and Millicell culture inserts (Millipore, Ireland, 6 slices per insert) were placed into 6-well plates (SPL Life Sciences Co., Pocheon, Korea) with 1 mL of Gahwiler's media per well. The slice culture media consisted of 25% Hank's Balanced Salt Solution (GibcoBRL/Life Technologies, Grand Island, NY, USA), 25% heat inactivated horse serum (Hyclone, Logan, UT, USA), 50% basal medium eagle (GibcoBRL/Life Technologies), 6.5-mg/mL glucose, and 200mM glutamax-I (GibcoBRL/Life Technologies). Slices were cultured at 37Ōäā with 5% CO2 in a humidified incubator (MCO175, Sanyo, Tokyo, Japan), and the media was changed twice per week. All procedures were performed aseptically in a horizontal flow hood.
2. Hypoxic injury
Slices were divided into two groups: 7 days in vitro (DIV) and 14 DIV. Hippocampal tissues at 7 DIV were representative of immature brains, while 14 DIV hippocampal tissues were representative of the mature brains14). The tissues in each group were exposed to 10% O2 for 60 minutes; deteriorating tissues were excluded prior to induction of hypoxic injury.
3. Image analysis
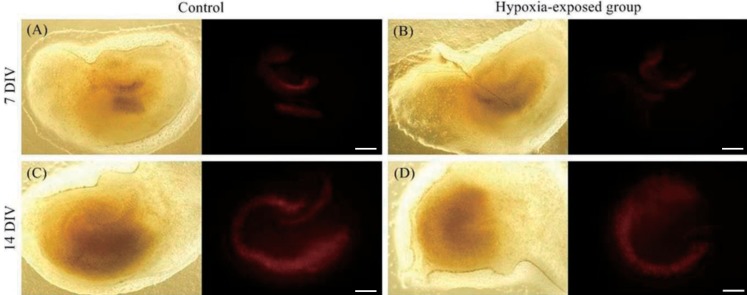
Fluorescent images of the slices were captured under an inverted microscope (IX 71, Olympus, Tokyo, Japan) 24 hours after hypoxic injury. To introduce total damage, slices were then exposed to 2mM N-methyl-D-aspartate (NMDA) in serum-free medium for 30 minutes at 37Ōäā with 5% CO2 in a humidified incubator. Slices were then cultured with 1 mL of serum-free media with 1-┬Ąg propidium iodide (Sigma Aldrich), and fluorescent images were obtained 24 hours after total damage. Images were analyzed with Image J (ver. 1.47c, National Institutes of Health, Bethesda, MD, USA), using a defined threshold as the amount of fluorescent area with a single researcher measuring individual areas above the threshold in the cornu ammonis 1 (CA1), CA3, and dentate gyrus (DG) (Fig. 1). After NMDA processing, images were assumed to include the total area of the hippocampal tissue. We then estimated the extent of damage 24 hours after hypoxic injury. Tissues in the 7 DIV and 14 DIV groups were compared, and hypoxia-exposed tissues were compared with the control group (Fig. 2). The ratio of damaged area at 24 hours to the total damaged area after NMDA treatment was also calculated.
Results
1. Cultured hippocampal tissues at 7 DIV
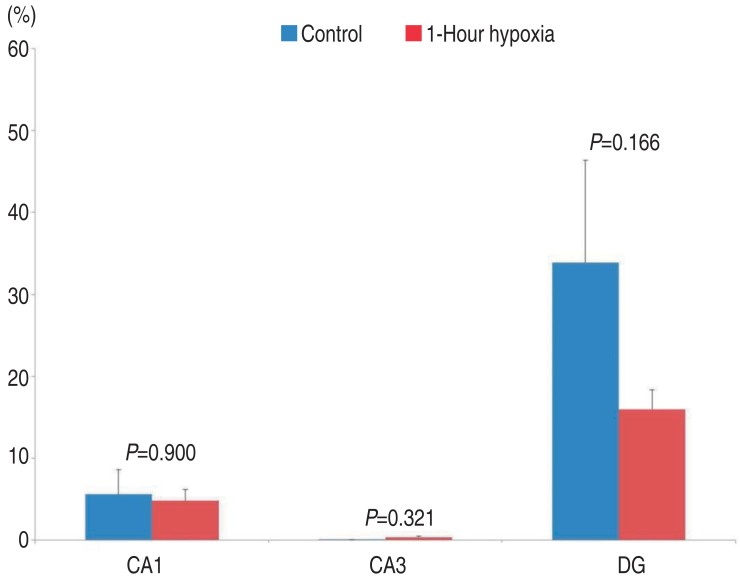
Damage to CA1 in hippocampal tissue at 7 DIV was 5.59%┬▒2.99% in the control group (mean┬▒standard error, n=30, measured area/total area). In tissues exposed to 10% O2 for 60 minutes, the damage was 4.80%┬▒1.37%. There were no significant differences between the tissues exposed to 10% O2 for 60 minutes and the control group (P=0.900).
Damage to CA3 in hippocampal tissue at 7 DIV was 0.04%┬▒0.02% in the control group. In tissues exposed to 10% O2 for 60 minutes, the damage was 0.35%┬▒0.13%. There were no significant differences between tissues exposed to 10% O2 for 60 minutes and the control group (P=0.321).
Damage to DG in hippocampal tissue at 7 DIV was 33.88%┬▒12.53% in the control group. In tissues exposed to 10% O2 for 60 minutes, the damage was 15.98%┬▒2.37%. There were no significant differences between tissues exposed to 10% O2 for 60 minutes and the control group (P=0.166).
In summary, compared to control group, in the 7 DIV group, tissues exposed to 10% O2 for 60 minutes tended to have decreased damage in the CA1 and DG (CA1: 5.59%┬▒2.99% vs. 4.80%┬▒1.37%, P=0.900, DG: 33.88%┬▒12.53% vs. 15.98%┬▒2.37%, P=0.166), but this decrease was not statistically significant (Fig. 3).
2. Cultured hippocampal tissues at 14 DIV
Damage to CA1 in hippocampal tissue at 14 DIV was 50.91%┬▒5.90% in the control group (mean┬▒standard error, n=30, measured area/total area). In tissues exposed to 10% O2 for 60 minutes, the damage was 32.30%┬▒3.34%. The tissues exposed to 10% O2 for 60 minutes showed significantly less damage than control tissues (P=0.004).
Damage to CA3 in hippocampal tissue at 14 DIV was 24.51%┬▒6.05% in the control group. In tissues exposed to 10% O2 for 60 minutes, the damage was 18.31%┬▒3.28%. There were no significant differences between tissues exposed to 10% O2 for 60 minutes and the control group (P=0.373).
Damage to DG in hippocampal tissue at 14 DIV was 15.72%┬▒ 3.47% in the control group. In tissues exposed to 10% O2 for 60 minutes, the damage was 9.91%┬▒2.11%. There were no significant differences between tissues exposed to 10% O2 for 60 minutes and the control group (P=0.134).
In summary, in the 14 DIV group, compared to the control group, tissues exposed to 10% O2 for 60 minutes showed decreased damage. This decrease was not significant in the CA3 (24.51%┬▒6.05% vs. 18.31%┬▒3.28%, P=0.373) and, DG (15.72%┬▒3.47% vs. 9.91%┬▒2.11%, P=0.134), but it was significant in the CA1 (50.91%┬▒5.90% vs. 32.30%┬▒3.34%, P=0.004) (Fig. 4).
Discussion
Asphyxia at birth is a frequent event that affects 4 million infants per year; moreover, about 1 million infants die from asphyxia while another 1 million experience persistent brain damage each year16). The causes of neonatal brain damage in response to hypoxic-ischemic insult are multifactorial17). In the developing brain, lack of oxygen availability results in an initial depletion of high energy phosphates, in particular adenosine triphosphate and phospho-creatine17).
The brain is particularly vulnerable to hypoxic conditions, such as those encountered in diving, high altitudes, aviation, and ischemia, especially hypoxia-ischemia followed by reperfusion18). Severe hypoxia-ischemia reperfusion leads to intracellular calcium overload, mitochondrial functional disorders, and structural lesions, resulting in a series of cellular functional impairments18). Moreover, mitochondrial dysfunction is involved in neuronal damage associated with hypoxic-ischemic reperfusion injury17). Recent studies strongly suggest the involvement of apoptosis in ischemia-induced delayed neuronal death19,20).
There are several in vivo and in vitro models that demonstrate the neuroprotective properties of sub-lethal hypoxia6,7,8,9,10,11,12). Gidday et al.10) first showed that a 3-hour pre-exposure of 8% oxygen to neonatal rat pups had protective effects over stroke produced by carotid occlusion and oxygen. Gorgias et al.12), investigated whether pretreatment with mild (15% O2) or moderate (10% O2) hypoxia could protect hippocampal neurons from damage induced by severe (3% O2) hypoxia. This group found that the moderate hypoxia pretreatment group had the most normal cells after severe hypoxia, as compared to mild hypoxia and control groups12). Xu et al.6), exposed organotypic hippocampal slice cultures to either OGD for 45 minutes (ischemia) or to OGD for 15 minutes (ischemic preconditioning) 48 hours prior to OGD. OGD was achieved by combining severe hypoxia with aglycemia6). Xu et al.6) found that there was significant protection by ischemic preconditioning when the delay between insults was 48 hours. Other in vitro models of ischemic preconditioning have provided robust neuroprotection11). For example, according to Khaspekov et al.11), an episode of 60 minutes of OGD, followed 1 and 2 days later by 90 minutes of OGD resulted in 40%-60% protection in mixed neuroglial hippocampal cell cultures. In their studies, OGD was achieved by glucose and argon-mediated oxygen deprivation11). Additionally, an in vitro study by Badaut et al.21), showed that when organotypic hippocampal slice cultures were exposed to a combined OGD of varying intensities, ischemic preconditioning was accomplished by exposure to the mildest ischemia condition (10% of O2 for 15 minutes) 24 hours before the severe deprivation (5% of O2 for 30 minutes). Moreover ischemic preconditioning not only prevented delayed ischemic cell death 6 days after insult but also the transient loss of evoked potential responses21).
The mechanisms of hypoxic and ischemic preconditioning are still debatable; both of these types of preconditioning appear to require RNA and protein synthesis10,22,23,24,25,26). Several molecules induced by hypoxia, such as erythropoietin and vascular endothelial growth factor are known to be protective27). Additionally, hypoxia-inducible factor (HIF) and HIF target genes are known to be related to hypoxia-induced tolerance28).
It has also been shown that mild to moderate hypoxia (>8% oxygen) does not produce neuronal cell death29,30). In order to determine the time of OGD exposure required to induce cell death, it was found that cultures subjected to 60 minutes of OGD did not increase cell death, while 90 minutes of OGD resulted in a significant increase in cell death11). Wise-Faberowski et al.31), evaluated the age-dependent effect of OGD in the developing rat brain by preparing organotypic hippocampal slice cultures from rat pups on postnatal days 4, 7, 14, and 21 and cultured at 7 DIV31). The slices were exposed to OGD for durations of 5-60 minutes and cell death was evident in organotypic hippocampal slice cultures prepared from post natal days 14 and 21 (P<0.001) with OGD durations of 5 and 10 minutes, respectively. As in these studies, percentage of oxygen, time of exposure for sublethal hypoxia, and the extent of sublethal hypoxia varied.
In our current study, the 60-minute duration of hypoxic exposure with 10% O2 was selected after reviewing previous articles and considering our experiences with organotypic hippocampal slice cultures. Interestingly, in hippocampal tissue at 14 DIV, the CA1 showed significantly decreased damage after exposure to 10% O2 for 60 minutes. Although it was not statistically significant, other tissues generally showed a tendency to have less damage after exposure to 10% O2 for 60 minutes.
These data indicate that the mature brain has a larger neuroprotective response to 10% O2 for 60 minutes exposure than the immature brain. Many mechanisms and molecules have been known to be associated with various types of preconditioning. These include HIF, lactate, adenosine A1 receptors, nuclear factor ╬║B, adenosine triphosphate-sensitive potassium channel, erythropoietin, vascular endothelial growth factor, and others27,28,32,33,34,35). The result in the current study that the mature brain exhibited a greater neuroprotective response than the immature brain, might be explained by the mechanisms and molecules associated with preconditioning; however, this will need to be explored further by future studies.
In relation to a clinical setting, our results might explain the reason why many neonates with HIE which had undergone similar hypoxic insults during labor result in different clinical outcomes. In other words, neonates most likely experience different levels of hypoxia during pregnancy so each case results in different neuroprotective effects on the brain.
In conclusion, our study suggests that mild hypoxia might have a neuroprotective influence in the brain. One limitation of our study was that we did not perform severe, lethal hypoxia to the mild hypoxia-exposed groups, so it was hard to correlate neuroprotective effects with ischemic preconditioning. Therefore, further studies are needed to determine the most effective protocol of generating a model of hypoxia-induced neuroprotection.


 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


