

Introduction
The reported incidence of tracheoesophageal fistula (TEF)/esophageal atresia (EA) is roughly 1-2 per 5,000 live births and can be seen regularly in most tertiary neonatal units that care for high-risk infants1). Since the first successful corrective surgery in 1939, the majority of neonates with TEF/EA can expect a satisfactory outcome due to the progress in surgery techniques, neonatal intensive care, and neonatal anesthesia over the past 60 years2, 3).
Herein, we report the clinical characteristics, combined anomalies of the TEF/EA, risk factors associated with mortality, and long-term morbidities of TEF/EA, according to Gross's4) anatomic classification, observed in a single neonatal intensive care unit over the past 18 years.
Materials and methods
1. Subjects
The subjects included those newly born infants with TEF/EA admitted from April 1994 to February 2007 who underwent corrective surgery at the neonatal intensive care unit of the Asan Medical Center. Patients transferred from other hospitals with suspected TEF/EA and whose diagnosis was later confirmed were included; however, patients transferred to the unit after corrective surgery was performed were excluded.
The medical records of 97 infants satisfying the above criteria were retrospectively reviewed for various prenatal and postnatal clinical characteristics, operative findings, combined anomalies, morbidities including esophageal strictures, gastroesophageal reflux, and weight gain for the 2-year period after birth. Various factors were reviewed to analyze the risk factors associated with the mortality of TEF/EF.
VACTERL association was defined if the patient has 2 or more anomalies of the vertebral, anorectal, cardiac (excluding patent ductus arteriosus, patent foramen ovale), renal/genitourinary, and limb systems.
2. Statistics
Multiple logistic regression analyses were performed on univariate variables, including hospital days, gestational age, birth weight, Apgar score, maternal age, polyhydramnios, type of TEF/EA, VACTERL association, ventilator days, time of operation, esophageal stricture requiring esophageal balloon dilatation under fluoroscopic guidance, and tracheostomy to seek risk factors associated with mortality. All data were considered statistically significant if P<0.05. SAS 9.1 (2003 version; SAS Institute Inc.; Cary, NC) was used.
Results
1. Prenatal characteristics
The average age (±standard deviation) of mothers who gave birth to newborns with TEF/EA was 30±3.8 years. Of the 91 (30%) mothers, 27 had , and prenatal diagnosis was suspected in only 6/50 (12%) (Table 1).
2. Characteristics of patients
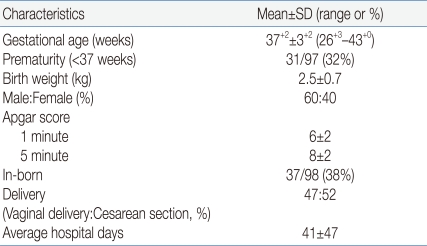
The average gestational age of newborns with TEF/EA was 37+2 weeks, and the percentage of premature infants was 32%. The average birth weight was 2.5±0.7 kg, and more males than females were diagnosed. The percentage of in-born newborns was only 38%. There was a slight preponderance of babies born via cesarean section, and the average length of hospitalization was 41±47 days (Table 2).
Of those newborns who underwent chromosomal analysis, 2 out of 19 had abnormal results: Edward syndrome (47,XX,+18) in 1 and DiGeorge syndrome (46,XX,t(4;5)(q35;q33.1) in another.
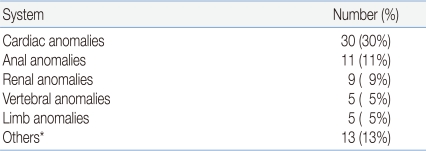
A total of 51% of the newborns had at least one congenital anomaly other than TEF/EA, and 41% of the anomalies were within the constellation of defects of VACTERL association. In order of approximate frequency, the affected organ systems most commonly associated with TEF/EA were cardiovascular, anorectal, genitourinary and skeletal. The most common congenital heart disease was tetralogy of Fallot (43%) followed by ventricular septal defect (20%), double outlet right ventricle associated with pulmonary atresia/pulmonary stenosis (13%), dextrocardia (10%), aortic stenosis and ventricular septal defect (3%), atrial and ventricular septal defect (3%), double outlet right ventricle and functional single ventricle (3%), and mitral atresia (3%). Of those with imperforate anus, 55% had high type.
The renal anomalies in the order of frequency were unilateral renal agenesis, vesicoureteral obstruction, horseshoe kidney, crossed renal ectopia, hydronephrosis, and multicystic dysplastic kidney. The vertebral anomalies, 5% of the total anomalies, included polydactyly, syndactyly, and club hand (Table 3).
Thirteen out of 40 neonates with VACTERL association had other congenital anomalies, including single umbilical artery (5 cases) followed by cleft palate (3 cases), craniosynostosis (1 case), agenesis of eyes and nose (1 case), small mandible (1 case), and tongue tie (1 case).
The gastrointestinal anomalies other than anorectal defects included duodenal obstruction, hypertrophic pyloric stenosis, malrotation, and biliary atresia. Congenital diaphragmatic hernia, subglottic stenosis, congenital dysplasia of hip, and hypoplastic thymus in a patient with DiGeorge syndrome were also observed in small numbers.
3. Surgery
Surgical intervention was performed, on average, the 4th day after birth on 81 neonates. Parents of 16 neonates refused further treatment due to the multiple congenital anomalies found. Immediate primary repair was possible in 72 cases (88%), and staged repair was performed in 8 cases (9%) after gastrostomy or duodenostomy due to the long esophageal gap. Simple ligation of fistula was done in 1 case (1%). The average length from the proximal to the distal esophageal pouch was 2 cm (range: 1.4-3 cm).
Out of 8 patients who underwent gastrostomy or duodenostomy, 4 patients expired preoperatively due to sepsis or respiratory failure from pneumonia before definitive corrective surgery. Two patients born in 1991 had esophageal replacement surgery with the colon at 14 months and 17 months of age, and 1 patient born in 2000 underwent delayed primary repair of the esophagus at 19 months of age.
The most common anatomical variant was type C, found in 71 patients (91%), followed by type A in 2 patients (2%), type E in 3 cases (4%), and type B or D in 1 case (1%). The average length of ventilation was 9.6 days (range: 1-106 days).
4. Postoperative complications
Among the 81 patients who underwent surgical corrections, contrast esophagogram was performed using barium on an average of 25 days after the surgery. Forty-seven patients (64%), 29 patients (39%), and 9 patients (12%) showed anastomotic strictures, reflux, and leaks, respectively. Of the 47 patients, 26 were demonstrated to have strictures on average 3 months after the surgery and required balloon dilatation an average of 1.3 times per patient. Recurrent fistulas occurred in 2 patients; primary surgical closure was required in 1, and spontaneous regression of fistula occurred in the other.
5. Mortality
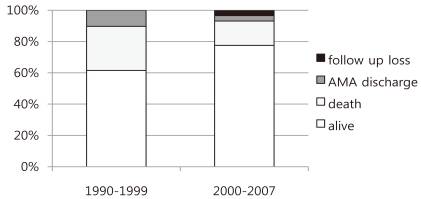
The study period was divided into 2 periods. The mortality rates for the periods 1990 to 1999 and 2000 to 2007 were 39% and 23%, respectively. Parents of 4 patients refused further treatment due to multiple congenital anomalies, resulting in 24 out of 39 patients surviving during the first study period in comparison to 2 patients whose parents refused treatment for the same reasons as in the first study period and 2 patients who were lost to follow-up, resulting in 45 out of 58 patients surviving during the second study period (Fig. 1).
Excluding those patients who did not get treatment and were lost to follow-up, the overall mortality rate was 20% (20/97 patients). The most common etiology causing death was sepsis followed by respiratory failure, subendocarditis, congestive heart failure, gastrointestinal hemorrhage, liver cirrhosis, and gastric perforation. Out of the 40 patients with VACTERL association, 30% (12/40 patients) died; the mortality rate was higher than the overall mortality rate. The mortality rate was 16% (5/30 patients) for those who had cardiac anomalies among patients with VACTERL association.
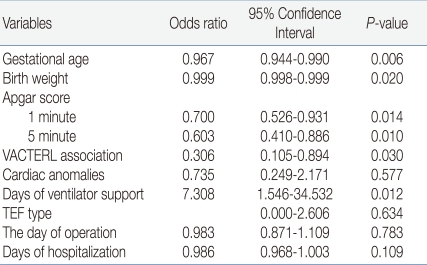
To assess the risk factors associated with mortality, multiple regression analyses of aforementioned univariate variables were conducted, and gestational age, birth weight, 1-minute and 5-minute Apgar scores, VACTERL association, and ventilator days were significantly associated with mortality (Table 4).
To delineate the effect of congenital cardiac anomaly and birth weight with prognosis, birth weight was stratified into 3 groups with or without associated cardiac anomalies: Out of 57, 45 infants survived. Of the 28 infants with cardiac anomalies and birth weight above 1,500 g, 67% survived; in contrast, 78% of those without cardiac anomalies and birth weight above 1,500 g survived (Table 5).
6. Morbidity
Excluding those who were lost to follow-up, a total of 72 patients were evaluated for respiratory and gastrointestinal morbidities. The morbidities included laryngomalacia (2%), cricopharyngeal incoordination (4%), recurrent pneumonia (4%), tracheostomy (5%), esophageal stricture, dysphagia (65%), and gastroenteral reflux (38%).
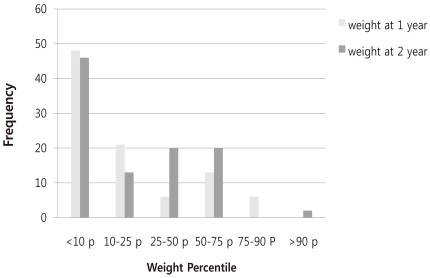
The weight gain at 1 year and 2 years of the patients who survived showed that 48% and 19% of the infants were below the 10th percentile and the 3rd percentile, respectively, at 1 year in contrast to 45% and 19%, respectively, at 2 years old (Fig. 2).
Of the 11 infants whose birth weight was below the 10th mesenchymal growth can stretch and disrupt the esophagus, creating esophageal atresia6).
Unlike other congenital anomalous disease, the prenatal detection rate of TEF is low: Sparey et al and De Vigan et al7, 8) reported that prenatal detection was possible in 9.2% and 24% of cases, respectively. The prenatal detection rate in our institution was 12%, a result reflecting more aggressive attempts to conduct prenatal diagnosis during the second phase of the study period. The positive predictive value of both polyhydramnios and a small or absent stomach bubble has been reported to be only 56%, making prenatal diagnosis even more difficult9).
The ratio of males to females was 6:4 in our study, a result similar to the incidence reported by Waterson et al10). Congenital malformations, including TEF/EA, are known to occur more commonly in twins. Mastroiacovo et al11) found that of 92 malformations studied, 39 malformations including TEF/EF were more common in twins than in singletons. The authors reported that the relative risk for TEF/EF in twins compared with singletons was 2.56 (95% confidence interval 2.01-3.25). In our series, the frequency of TEF/EA among twinning was 5%, and of interest, TEF/EF was observed in both fraternal twins.
The incidence of other anomalies associated with TEF/EA is reported to be 30-60%10, 12, 13), a result similar to our study of 52% where one or more anomalies was found. The most common anomaly was cardiovascular (35%) followed by genitourinary (24%), gastrointestinal (24%), musculoskeletal (13%), and neurologic (10%). An abnormal chromosomal study was found in 10% in our study, a result similar to other reports7, 14, 15). The most commonly occurring congenital heart disease as reported by Choi et al3), in order of frequency, was ventricular septal defect, patent ductus arteriosus, atrial septal defect, aortic stenosis, tetralogy of Fallot, truncus, and transposition of great vessels, whereas our finding suggested that tetralogy of Fallot was the most commonly found congenital heart disease. About 1/3 of patients presenting with a gap of more than 2 cm between the proximal and distal esophagus were reported to result in anastomotic leak, recurrence of TEF, gastroesophageal reflux, and anastomotic stricture as postoperative complications3, 16). An ultra-long gap of more than 3 cm was more frequently observed in patients with vascular ring, TEF with proximal esophageal atresia, or esophageal atresia without TEF3, 16, 17) in which frequent postoperative complications, including respiratory compromise, dysphagia, and growth failure18), are known to occur. This was also true in our study of 7 infants with an ultra-long gap; 4 infants died, 2 infants showed failure to thrive, and only 1 infant showed normal growth for age.
The 20% mortality rate in our study is similar to the mortality rates reported by others of 10-30%12, 13, 19). Variables, including gestational age, birth weight, 1-minute and 5-minute Apgar scores, VACTERL association, and duration of ventilator days, seemed to affect the mortality rate in patients with TEF/EA, a finding different from that reported by Choi et al3). Nevertheless, both reports are in agreement in that prematurity is an important variable influencing survival: a survival rate of 87% among full-term infants in comparison to a 62% survival rate among preterm infants. Boyle et al17) and Spitz et al20) reported that the survival of full-term infants with TEF/EA without associated anomaly was 95-100%.
In contrast to reports by Sparey et al7) and Engum et al21), our study showed that gastrointestinal complications were more frequently observed than respiratory-related complications. Esophageal stricture and gastroesophageal reflux were observed in 65% and 39%, respectively, of our study population.
The upper gastrointestinal morbidity and anthropometric data collected by Chetcuti and Phelan22) in 334 patients aged 1 to 37 years with repaired TEF/EA showed that 67% required hospitalization with esophageal complications and half underwent one or more surgical procedures. More importantly, the patients' median weight centile was the 25th. Such findings and a report by Kim et al23) are in accordance with our report in which the percentage of infants whose weight remained below the 10th percentile at 1 year old and 2 years old was 48% and 45%, respectively.
In conclusion, the overall mortality rate (20%) and morbidity rate (67%) of infants born with TEF/EA are high due to the associated anomalies and gastroesophageal and respiratory complications during infancy. Longer follow-up into childhood and adulthood is needed in the future as the long-term outcome of the majority of adults who experienced a difficult initial period is reported to be excellent, and they enjoy a normal lifestyle22).


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


