

Introduction
Loeys-Dietz syndrome (LDS) is a recently recognized genetic aortic aneurysm syndrome characterized by the triad of hypertelorism, bifid uvula or cleft palate, and generalized arterial tortuosity with aneurysms and dissections throughout the arterial tree1). LDS is associated with mutations in the transforming growth factor beta receptor type I (TGFBR1) and type II (TGFBR2) genes1,2). These mutations cause increment of downstream TGFB signaling in the aortic media and subsequent overproduction of collagen, disarrayed elastic fiber and loss of elastin content in extracellular matrix1-3). Therefore, patients with LDS usually show aggressive and rapid progression of aortic dilatation and regurgitation, and have a high risk of aortic dissection or rupture, even though the patients are young in age and had smaller aortic diameters than other aortic aneurysm syndromes.
We report a child who was diagnosed as LDS with specific, characterized phenotype and two novel gene missense mutations in TGFBR2 gene, and underwent surgical repair for aortic root aneurysm.
Case report
A 7-year-old girl was consulted from another department because of an abnormal electrocardiogram (ECG) pattern of severe left axis deviation and tall R in V5-6, and cardiomegaly (cardiothoracic ratio 0.62) in a chest X-ray. In physical examinations, she had ocular hypertelorism, bifid uvula (Fig. 1A) and high arched palate, strabismus, pectus excarvatum, arachnodactyly, calcaneus eversion and metatarsus adductus (Fig. 1B, C).
Weight and height were in the 30th and >97th percentile, respectively.
In past history, she was born at term to a 30-year-old mother and a 30-year-old unrelated father. Her mother's obstetric history was gravida 4, para 1, abortion 3 and was unremarkable. She was referred to our neonatal intensive care unit on day 1 of life for additional evaluation of a genetic syndrome in the setting of diffuse hypotonia and musculoskeletal abnormalities. All growth parameters including height, weight and head circumference were within normal limits. Family history was unremarkable. Investigations included chromosomal study, skeletal imaging, ultrasonogram of the head, and echocardiography. Although she had abnormal skeletal morphology, her chromosome was normal. She had no intracranial abnormalities. A small patent ductus arteriosus without aneurysm and a small atrial septal were defected on echocardiography.
She underwent serial orthopedic surgical intervention for correction for lower-extremity abnormalities at neonatal and childhood period and had done regular follow-up at the orthopedic department. The patient was subsequently lost to the cardiology department follow-up until cardiology consult was performed because of cardiomegaly and abnormal ECG findings for ophthalmologic intervention due to strabismus.
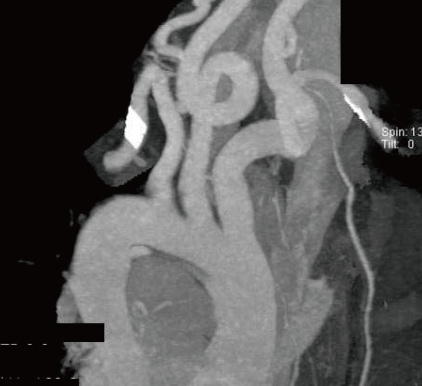
When she was consulted for abnormal ECG, subsequent echocardiography was performed and revealed marked dilated aortic annulus and root4), which measured 23 to 24 mm and 33 to 35 mm, respectively (Z-value >2, body surface area =0.83), with grade II aortic regurgitation, dilated pulmonary annulus (24 to 25 mm, Z-value >2), and small cone shaped patent ductus arteriosus. Computed tomography (CT) angiography showed arterial tortuosity at the common carotid artery (Fig. 2).
Based on above findings, the suspicion of LDS rather than Marfan syndrome (MFS) was raised because she had all of the typical triad of LDS, such as facial abnormalities (hypertelorism and bifid uvula), markedly dilated aortic annulus and root in spite of young age and arterial tortuosity of the neck vessels. Moreover she did not have ectopia lentis or myopia which is the frequent findings in MFS.
To confirm our clinical diagnosis of LDS, we performed molecular genetic testing of the TGFBR1 and TGFBR2 genes. Informed consent of the parents was obtained prior to genetic testing.
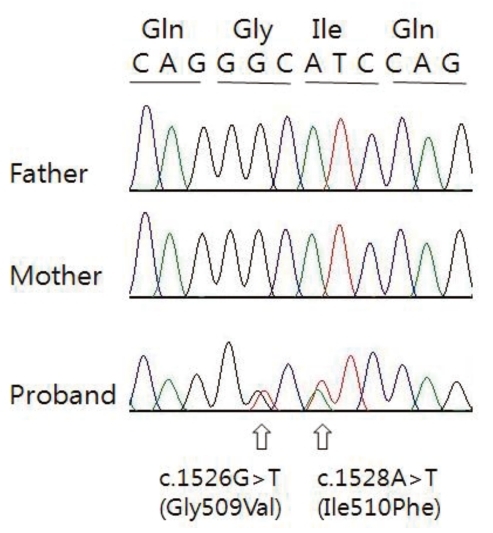
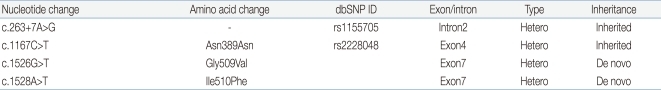
All coding exons and flanking intron regions of the TGFBR1 and TGFBR2 genes were amplified and sequenced using primer sets designed in our laboratory. In TGFBR1 gene, we could not detect any variations, whereas we identified one synonymous, one intronic and two missense variations in the TGFBR2 gene (Table 1). Of these, one intronic (c.263+7A>G, rs1155705) and one synonymous (c.1167C>T, rs2228048) variations were inherited from one of parents and known-polymorphisms listed in the single nucleotide polymorphism database (dbSNP; http://www.ncbi.nlm.nih.gov/projects/SNP/). However, the two heterozygous missense variations (c.1526G>T c.1528A>T) were novel variations which have not been described in any previous literatures. These two missense variations were not detected in the proband's parents suggesting as de novo (Fig. 3). These variations are located in exon 7 and are included in the highly conserved serine/threonine kinase domain XI of TGFBR2. Furthermore, when we checked the influence of these variations to the function of protein using sorting intolerant from tolerant algorithm, both of those variations are expected to affect protein function. When we used the polymorphism phenotyping (PolyPhen) algorithm, Gly509Val was expected as 'probably damaging variation' and Ile510Phe was expected as benign one.
Medication of angiotensin receptor II antagonist (losartan, 0.5 mg/kg/day) was started and prophylactic surgical repair for aortic root aneurysm such as valve sparing root replacement was performed. So far, surgical intervention was successful, she has had a medical checkup at regular intervals with losartan medication and CT angiography every year.
Discussion
Loeys et al.2) first reported six families who had phenotype characterized by typical cardiovascular (generalized arterial tortuosity and aneurysms with dissection throughout the arterial tree), craniofacial (hypertelorism, bifid uvula and/or cleft palate), and skeletal (pectus excarvatum, dolichostenomelia, arachnodactyly and metatarsus adductus) manifestations, and heterozygous mutations in the genes encoding TGFBR I or II. They described this phenotype as LDS, a new aortic aneurysm syndrome. So far, more than 80 LDS patients including some pediatric patients have been described in previous papers, and many TGFBR1 or TGFBR2 gene mutations have been also reported in those patients2,5-7).
TGFBR2 gene, which is located chromosome 3p22.5, consists of seven exons and six introns, and encodes the human TGFBR II8). This receptor regulates cellular proliferation, differentiation, motility, organization, apoptosis, and formation of extracellular matrix, especially in the cardiovascular system9,10). Mutations of TGFBR2 gene are associated with increased downstream TGFB signaling in the aortic media, overproduction and deposition of collagen, organization of elastic fiber and loss of elastin content in extracellular matrix1-3). Excessive collagen deposition results in weakness of aortic vascular bed, dilatation and dissection of the aorta.
The LDS phenotype may resemble that of the MFS. MFS is characterized by skeletal, ocular, cardiovascular, pulmonary, skin findings, and dural ectasia. Among of these findings for MFS, specific ocular finding, bilateral ectopia lentis occurs in about 40 to 56% of patients with MFS11) and does not occur in LDS1). In comparison to MFS, LDS patients show typical characteristics such as facial dysmorphology (hypertelorism, bifid uvula and/or cleft palate), aortic root aneurysm, aneurysm of other vessels and widespread arterial tortuosity1,2,12). If the patient has typical characteristics for LDS and does not have ectopia lentis, the patient can be diagnosed with LDS and gene testing for LDS should be performed.
In respect to specific genotype, LDS patients show TGFBR1 or TGFBR2 gene mutations but do not show mutations in the gene encoding fibrillin-1 (FBN1). On the other hand, most of MFS patients show mutations in the gene encoding FBN1 although some of MFS patients have been reported to have TGFBR mutations without FBN1 mutations5). LDS patients have arterial tortuosity and aneurysms throughout the arterial tree, whereas the main target vessel in MFS is the ascending aorta and aortic root1,2,12). Therefore, the initial evaluation of patients with a presentation similar to that of MFS requires a multi-disciplinary approach including clinical genetics, cardiology, ophthalmology and radiology.
The most important finding of LDS is the aggressive and rapid progression of aortic pathology even though the patients are young in age and shorter median survival due to occurrence of dissections at smaller diameters than in other connective-tissue disorders1,13). The median survival was 37 years among patients with LDS1), 48 years among patients with vascular Ehlers-Danlos syndrome14) and 70 years among patients with MFS who underwent treatment15). In previous report, mean age of operation was 9.2+5.7 years (range, 0.5 to 17 years) in pediatric patients undergoing aortic surgery13). Among 14 pediatric patients, 3 patients aged younger than 10 years had fatal aortic dissection and intracerebral hemorrhage, and these findings occurred in patients who had a smaller aortic root diameter than in MFS patients13). Our patient also showed marked aortic root dilatation and progressive aortic regurgitation when she was 7-year-old, so we considered early surgical intervention.
In conclusion, patients with distinct phenotypic characteristics, marked aortic dilatation and aneurysm at early age should be suspected to be affected by LDS and could benefit from rapid TGFBR1 or TGFBR2 gene analysis. Early genetic diagnosis is the essential tool to make adequate management for LDS patients. If clinical diagnosis is confirmed by molecular genetic testing of the TGFBR genes, according to guideline13), medical checkup and clinical assessments using echocardiography, CT or magnetic resonance imaging angiography at regular intervals, and early, prophylactic surgical intervention of abnormal aortic dilation and aneurysm such as valve sparing root replacement, must be done to prevent future vascular events.

 PDF Links
PDF Links PubReader
PubReader PubMed
PubMed Download Citation
Download Citation


