


Introduction
The anthracycline antibiotic adriamycin (ADR) is considered a very effective antitumor agent which is used in the treatment of solid tumors and malignant hematological disease such as leukemia, lymphomas and many solid tumors1).
However, there is an important limitation to the use of ADR, which is dose-related cardiotoxicity2). The acute side effects are myelosuppression, nausea, vomiting and arrhythmias3). These can occur immediately after treatment and they are characterized by transient arrhythmias, pericarditis and reversible hypotension. But, chronic side effects such as cardiomyopathy and congestive heart failure (CHF) are irreversible, and, they have grave prognosis4). Chronic toxicity, usually caused by doses above 550 mg/m2, may cause CHF5).
Multiple mechanisms have been proposed to explain the development of ADR-induced cardiomyopathy. They include free radical formation6), a reduction in myocardial antioxidant enzyme activities7), inhibition of the carnitine palmitoyl transferase system I8), lipid peroxidation3), the inhibition of nucleic acid and protein synthesis1), abnormalities in the mitochondria9), imbalance in myocardial electrolytes1) and apoptosis10).
Mitochondrial damage is considered to be at the forefront of the pathogenesis of ADR cardiomyopathy because the onset and severity of cardiomyocyte injury correlates with mitochondrial radical oxygen species (ROS) production and the disruption of bioenergetics11). Thus, although mitochondria are important in ADR-induced apoptosis, myofibrillar deterioration, and intracellular calcium dysregulation are also important mechanisms associated with ADR-induced cardiac toxicity11).
ADR-induced apoptosis1) and hyperlipidemia8) may also be involved in the process. Programmed cell death, or apoptosis, has been put forth as being involved in cardiac dysfunction under some experimental and clinical conditions12). Not only cardiomyocytes but endothelial cells are also affected, as indicated by caspase activation and inter-nucleosomal DNA degradation. It is commonly accepted that the oxidative stress induced by ADR activates apoptotic signaling leading to cardiomyocyte apoptosis13), and that both the extrinsic and intrinsic apoptotic-pathways are involved14).
Increased immuno-reactive caspase-3 expression was found in ADR-treated rats. Several studies have shown a dose-related increase in apoptosis by flow cytometry, DNA ladder analysis, terminal deoxyuridine triphosphate nick end labeling (TUNEL) assay and electron-microscope examination2). ADR-induced apoptosis has been associated with multiple signaling pathways15). The expression of Bcl-2 was down-regulated and that of caspase-3 was up-regulated in the ADR induced apoptosis of both cardiomyocytes and endothelial cells2). Bax expression was increased in the cardiomyocytes but unchanged in the endothelial cells2).
Remodeling of extracellular collagen matrix plays a major role in left ventricle (LV) hypertrophy. Fibrosis has been suggested to be involved in cardiac stiffness and dysfunction, which are caused when an increase in collagen synthesized by the fibroblasts invades and replaces necrotic or apoptotic myocytes16). Significant alterations in the structure and composition of the extracellular matrix contribute to the development of heart failure17). Both matrix metalloproteinase (MMP)-2 and MMP-918) are believed to contribute to cardiomyopathy by weakening the collagenous matrix against which the cardiomyocytes work.
The purpose of this study was to investigate changes in several gene expressions associated with apoptosis and remodeling in ADR-induced cardiomyopathy rat models.
Materials and methods
1. Animals
Sprague-Dawley rats between 8 and 12 week age, weighing 300ŌĆō450 g were used. All animals were housed in a temperature-regulated room. Groups were divided into 2 groups: the control (C) group and the ADR (A) group. The A group was injected ADR 5 mg/wk for 3 weeks (intraperitoneal injection). We sacrificed the 6 rats in each group at 3 weeks.
This study was approved by the Institutional Animal Care and Use Committee at Ewha Womans University (approval number: 15-0319).
3. Measurement of body and organ weight
While the rats were anaesthetized with zoletil (Virbac, Carros, France) and rompun (Bayer, Seoul, Korea), organs were immediately removed and weighted. The hearts were excised and a portion of the LV was rapidly frozen in liquid nitrogen and stored at ŌłÆ70Ōäā for studies of western blot analysis. LV hypertrophy were calculated LV+septum (S)/right ventricle (RV).
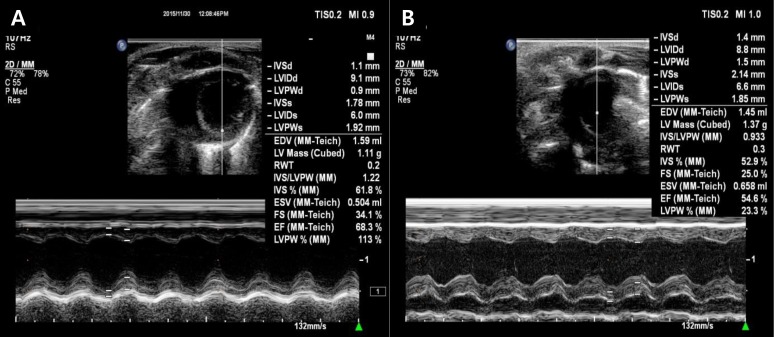
4. Echocardiographic parameters
Echocardiography was performed using an IE33 machine (Philips Medical System, Andover, MA, USA) with an S12 transducer at week 3. M-mode and 2-dimensional echocardiography studies were performed. Standard parasternal and apical views were acquired.
5. Western blot analysis in the LV tissues
LV tissues were homogenized in 4Ōäā lysis buffer (PRO-PREP, iNtRON Biotechnology, Seongnam, Korea). The homogenate was centrifuged at 12,000 rpm for 30 minutes. Protein concentrations were quantified by bicinchoninic acid assay (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of total proteins were loaded electrophoresed and transferred to nitrocellulose membranes. The membranes were blocked with 5% bovine serum albumin in tris buffered saline with 0.1% tween 20 (TBST) for 1 hour and washed 4 times with TBST every 5 minutes. Primary antibodies were incubated 4Ōäā overnight and secondary antibodies including caspase-3, Bax (Abcam, Cambridge, United Kingdom), interleukin (IL)-6, tumor necrosis factor (TNF)-╬▒, brain natriuretic peptide (BNP), collagens 1 and 3 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Abclon, Seoul, Korea), at 4Ōäā overnight were incubated for 1 hour at room temperature. The membranes were developed using luminescence detected by LAS-3000 (Fuji Film Corp., Tokyo, Japan) using enhanced chemiluminence reagent (Amersham Corp., Arlington Heights, IL, USA). The expression of GAPDH was used as an internal standard.
6. Masson's Trichrome staining in the LV tissues
Hearts were removed and fixed with 4% neutral phosphate buffered formalin. Paraffin embedded sections of hearts were stained by Masson's Trichrome staining method using standard procedures.
7. Terminal deoxyuridine triphosphate nick end labeling staining assay in the LV tissues
TUNEL assay was used for the visualization of apoptotic cells using a TUNEL apoptosis detection kit (Apoptag, Millipore, MA, USA). The TUNEL assays were performed according to the manufacturer's instruction. Sections were deparaffinized, rehydrated and incubated with proteinase K (20 mg/mL). After an application of blocking solution (5 minutes, RT), avidin-fluorescein isothiocyanate (FITC) was applied on slides for 30 minutes at 37Ōäā. Samples were mounted using mounting medium (Dako, Carpinteria, CA, USA) with propidium iodide.
Results
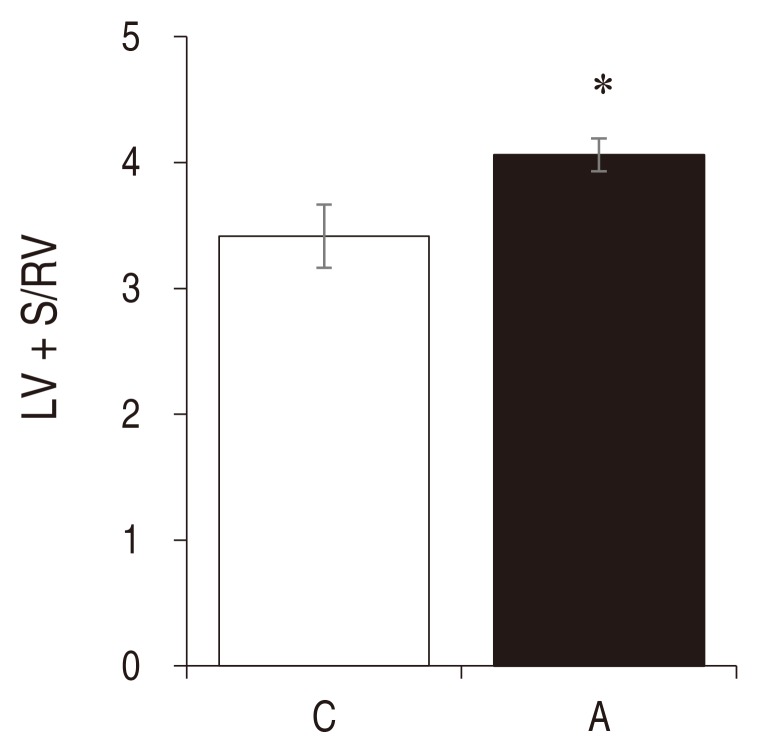
2. LV hypertrophy after ADR injection
An index of LV hypertrophy, LV+S/RV ratio was significantly increased in the A group compared with the C group (C vs. A: 3.42┬▒ 0.25 vs. 4.06┬▒0.13 , P<0.05 ) (Fig. 2).
3. Increased collagen contents after ADR injection
As remodeling of extracellular collagen matrix plays a major role in LV hypertrophy, collagen 1 (Fig. 3A) and 3 protein expression levels (Fig. 3B) were significantly increased in the A group compared with the C group. Collagen contents was significantly increased in the A group compared with the C group (52.42┬▒3.41 vs. 80.45┬▒7.43, P<0.05) (Fig. 3C).
Increased myocardial collagen contents by ADR may affect LV stiffness and dysfunction.
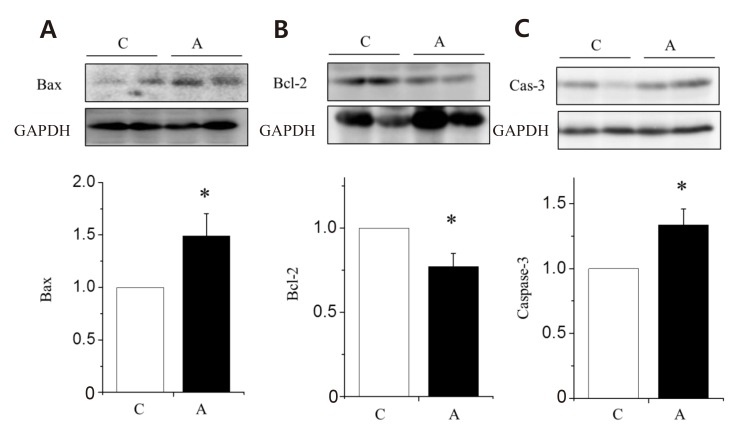
4. Changed apoptotic protein expression levels after ADR injection
To find out which pathways are involved in ADR-induced LV remodeling, we investigated the changes of apoptotic protein expressions in the LV tissues. The apoptotic protein, Bax (Fig. 4A) and caspase-3 (Fig. 4C) were significantly increased in the A group compared with the C group. Antiapoptotic protein, Bcl-2 was significantly decreased in the A group compared with the C group (Fig. 4B).
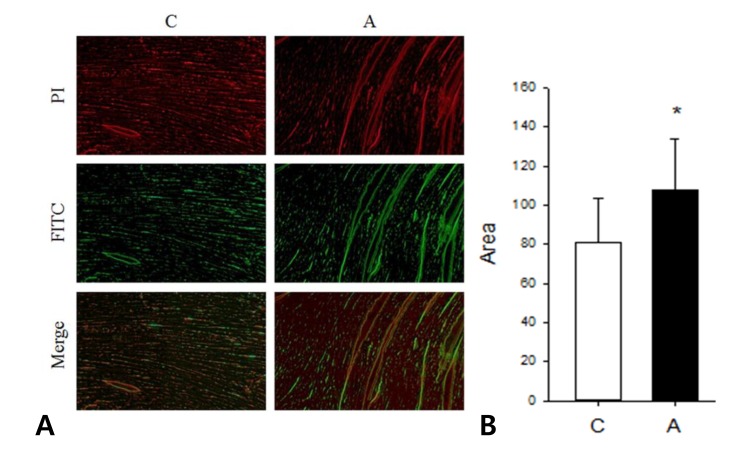
6. Increased apoptosis after ADR injection by TUNEL assay
We investigated apoptotic area in the LV tissues of ADR injected rat by TUNEL assay. Apoptosis was significantly increased in the A group compared to the C group (C vs. A; 80.79┬▒23.00 vs. 108.11┬▒ 25.82, P<0.05) (Fig. 7).
Discussion
An important finding of this research was increased apoptosis in the myocardium of the ADR-treated rats. We wanted to find out which pathways were involved in ADR-induced LV remodeling, so we investigated changes of apoptotic protein expressions in the LV tissues. We found that the apoptotic proteins such as Bax and caspase-3 were significantly increased in the A group compared with the C group. In addition, antiapoptotic protein, Bcl-2 was significantly decreased in the A group compared with the C group. The use of rats in animal models has provided valuable information for understanding the pathogenesis of this form of cardiomyopathy.
Wu et al.2) reported an increase in Bax and caspase-3 gene expressions and a decrease in the expression of Bcl-2 in the ADR-treated cardiomyocytes using northern blot analysis, and reverse transcriptase polymerase chain reaction. These data were similar with our data and it suggests that cardiomyocyte cell apoptosis may play an important role in ADR cardiomyopathy.
There are some controversies between the correlation of dose of ADR and apoptosis. Recent reports have described cardiomyocyte and endothelial cell death through apoptotic mechanisms in a variety of cardiovascular diseases and pathophysiological states19). To confirm the presence of apoptosis, several different detection techniques were used, including DNA ladder analysis, flow cytometry and TUNEL assay2). Arola et al.20) demonstrated an increase in cardiomyocyte apoptosis with dose of ADR, even after a single intraperitoneal injection of 2.5 or 5 mg/kg. Zhang et al.21) showed endothelial cell apoptosis in the absence of clinical signs of CHF following intravenously injection of ADR at up to a total of 12 mg/kg over a 12-week period.
ADR alone cannot completely explain the extent of apoptosis in ADR-treated animals who suffer heart failure. Using a single injection of 2.5 or 5.0 mg/kg of ADR, Arola et al.20) determined that the percentage of cardiomyocyte apoptosis peaked on the first day after injection, and declined thereafter. Repeated injections of 2.5 mg/kg of ADR resulted in apoptotic peaks 24 hours after each injection. However, a return to baseline levels occurred within a week of treatment. These results suggest that, at each single dose of 2.5ŌĆō5 mg/kg of ADR administration, acute cardiotoxicity with cardiomyocyte apoptosis happens only temporarily.
On the contrary, Zhang et al.21) did not observe any cardiomyocyte apoptosis in ADR-reated rats at a cumulative dose of 12 mg/kg. Nakamura et al.22) administered ADR to male Wistar rats via a tail vein at incremental dosage of 2 mg/kg weekly and found no evidence of cardiomyocyte apoptosis and no clinical evidence of heart failure up to even high doses of 16 mg/kg. Cardiomyocyte apoptosis increased significantly, but only at cumulative doses of 18 and 20 mg/kg of ADR and occurred together with pleural effusion, indicating heart failure. This suggests that in addition to transient cardiomyocyte apoptosis occurring with each single-dose injection of 2ŌĆō5 mg/kg of ADR, sustained cardiomyocyte apoptosis together with clinical signs of CHF also occurs in cumulative ADR doses of 15 mg/kg or more.
In our study, increased apoptotic proteins such as Bax and caspase-3 and decreased antiapoptotic protein such as Bcl-2 were noted with a cumulative ADR dose of 15 mg/kg in the A group compared with the C group. Heart failure such as tachypnea, pleural effusion and ascites was also observed in the A group.
Even though cardiomyocyte and endothelial cell apoptosis have been reported in humans and experimental animals with dilated cardiomyopathy and heart failure2), the relationship between apoptosis and CHF has not been fully elaborated. Some investigators have suggested that CHF itself might induce cardiomyocyte and endothelial cell apoptosis23).
Caspase activity can also be influenced by ADR. Apoptosis is associated with ADR administration in vivo, but the results have not been duplicated in isolated cardiomyocytes24). It is difficult to determine whether or how ADR directly influences caspase activity, as many pathways can contribute to the activation of caspase-dependent apoptosis25).
After doxorubicin exposure, apoptosis is started by both caspase-3 activation. There is increasing evidence that apoptosis contributes substantially to the pathogenesis of CHF.
It reduces the number of functioning contractile cardiomyocytes. Importantly, apoptosis of nonmyocytes can also have a negative effect on the failing myocardium, contributing to adverse ventricular remodeling, and playing an important role in the transition to an end-stage decompensated stage, regardless of the underlying etiology of CHF26).
The remodeling of extracellular collagen matrix plays a major role in LV hypertrophy. It has been suggested that fibrosis is involved in cardiac stiffness and dysfunction, caused by an increase in collagen synthesized by the fibroblasts which invades and replaces necrotic or apoptotic myocytes16). In our study, collagen contents in the LV tissues were significantly increased in the A group compared with the C group. Increased myocardial collagen contents by ADR might affect LV stiffness and dysfunction.
Both cellular and extracellular factors have a role in the complex process of myocardial remodeling. Significant alterations in the structure and composition of the extracellular matrix contribute to the development of heart failure17). ADR has the opposite effect on the heart, enhancing a production of MMP-2 and -9. Both MMP-2 and MMP-918) are thought to contribute to cardiomyopathy by weakening the collagenous matrix against which the cardiomyocytes work and contribute to pathological remodeling. Both MMP-2 and MMP-9 activities are enhanced by ADR-induced ROS generation10).
In our study, LV+S/RV ratio significantly increased in ADR treated rats. It suggests LV hypertrophy. Fractional shortening and ejection fraction were significantly decreased by echocardiography after ADR treatment. Serial echocardiographic monitoring is generally used for cardiotoxicity detection. Evaluation of LV systolic function using ejection fraction, or fractional shortening by echocardiography can be used to detect the development of cardiomyopathy. However, these are insensitive and still inaccurate markers of early ADR injury, as the guidelines for terminating ADR27).
Since ADR disrupts cardiac myocyte cell membranes, biomarkers can be used to assay for the presence and extent of myocyte injury. Lipshultz et al.28) reported the efficacy of cardiac troponin T as a possible quantifier for acute ADR-induced myocardial injury. Other potential markers include plasma levels of circulating natriuretic peptides, such as atrial-type natriuretic peptide and BNP, which are elevated in LV dysfunction and heart failure. Levels of these proteins were significantly elevated in patients treated with ADR who had cardiac dysfunction, compared with healthy controls or patients with normal cardiac function10).
Cardiac biomarkers can be good indicators of ADR-induced myocardial injury and can provide useful diagnostic information, especially when used in combination with assessment of LV function. In our study, BNP protein expressions in LV the tissues were significantly increased in the A group compared with the C group.
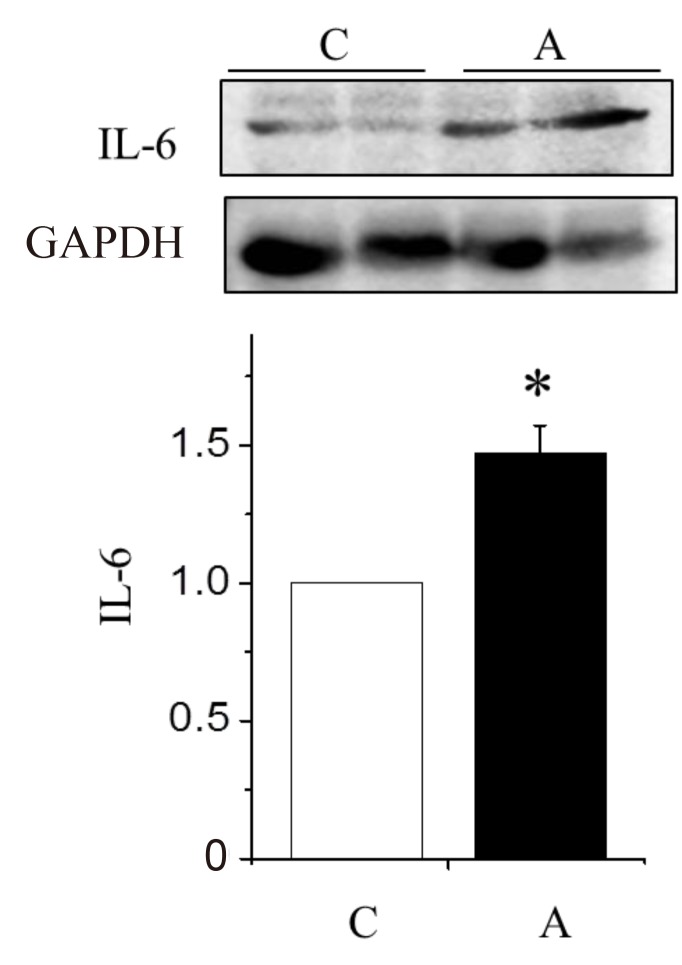
Several candidate mediators such as IL-1╬▓, TNF-╬▒ elevated in serum levels during CHF, can induce cardiomyocyte apoptosis in vitro29). In our study, IL-6 protein expressions in the LV tissues were significantly increased in the A group compared with the C group.
Cesselli et al.30) have demonstrated that apoptosis of cardiomyocytes, endothelial cells and fibroblasts occurred before any clinical signs of CHF. It appears likely, therefore, that cardiomyocyte apoptosis may be associated with decreased myocyte numbers, and with replacement fibrosis. Endothelial cell apoptosis may also reflect the disappearance of cardiac capillaries, and a regional reduction in blood flow and decreased tissue oxygenation. Importantly, induction of these changes precedes the development of LV dysfunction, providing strong additional support for the proposition that apoptosis is involved in LV remodeling29).
The limitation of this study is the small number of experiments.
In conclusion, the results of this study indicate that cardiomyocyte death can occur via apoptosis, with the dose-related induction occurring with changes in the expression of the apoptosis-related genes such as caspase-3, Bcl-2, and Bax expression. These results provide evidence that apoptosis may play an important role in ADR-induced cardiomyopathy.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


