




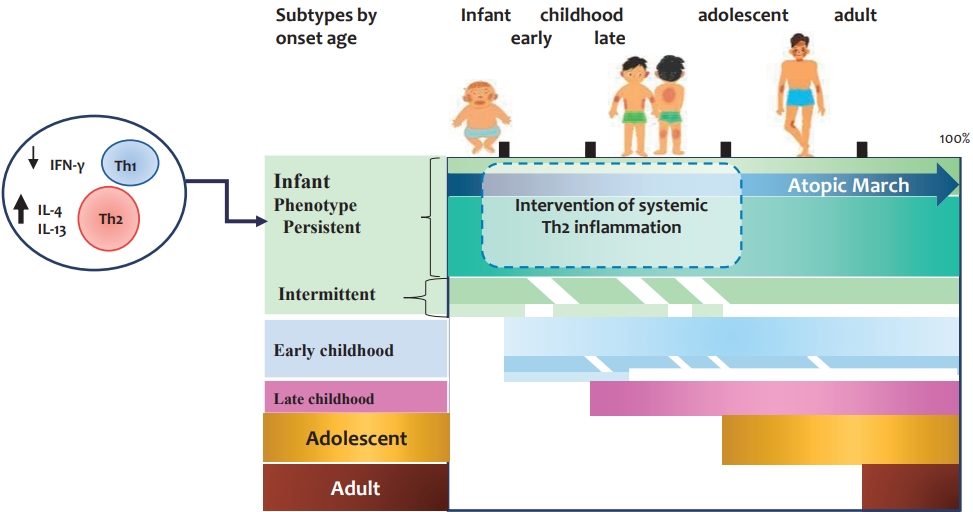
Graphical abstract. Subtypes of adults with atopic dermatitis. Approximately 40%–60% of adults with atopic dermatitis develop them in infancy and persist into adulthood. At birth, reduced IFN-γ- and enhanced IL-4-producing CD4+ cord blood T cells are subsequently associated with infancy-onset atopic dermatitis. Early-onset atopic dermatitis and allergic sensitization at early age increase the risk of the early-onset persistent phenotype. Among infants with early-onset atopic dermatitis, some develop systemic allergic disease such as food allergy, allergic rhinitis, and asthma. The figure is based on the findings described by references 8–13, 15, and 16. IL, interleukin; IFN, interferon; Th1, T helper 1; Th2, T helper 2.
Introduction
Atopic dermatitis (AD) was once considered an early-onset pediatric disease that usually resolves around the age of 2–3 years [1]. Although approximately 40%–60% of infancy-onset AD cases achieve remission by 6–7 years of age, recent studies reported that AD is a lifelong disease with recurrent exacerbations [2-4]. AD is frequently accompanied by systemic allergic diseases, other inflammatory diseases, and/or psychosocial disorders such as depression, anxiety, sleep disorders, and attention deficit hyperactivity disorder [5,6]. Therefore, the burden of AD is quite high. Given the fact that 85% of all AD cases begin within the age of 5 years, this period may be critical not only for AD development but also for disease modification.
Recent studies have provided new insights into the complex pathophysiology and phenotypes of AD. Moreover, based on an extended understanding of its pathogenesis, new agents are being tested in patients with moderate to severe AD. This review describes the latest knowledge about pediatric AD, the pathogenesis and phenotypes of it, and the currently available systemic therapeutics for children with moderate to severe AD that is refractory to topical treatment.
Atopic march begins with atopic dermatitis
“Atopic march” is the progression of allergic diseases from AD to other immunoglobulin E (IgE)-mediated diseases such as food allergy (FA), allergic rhinitis, and asthma. Over the past 20 years, the term atopic march has been widely used to describe changes in the temporal prevalence of allergic diseases reported in epidemiological studies ranging from AD and FA in infancy to allergic rhinitis and asthma in childhood [7]. These results led to the hypothesis that AD is the first manifestation of an atopic phenotype starting in infancy and early childhood [8]. There is epidemiological and experimental evidence supporting AD as the initiation of allergic diseases [8,9]. Several prospective birth cohorts have shown an association between early-onset AD and the development of asthma and allergic rhinitis at school age [8-10]. The risks of respiratory allergic diseases are greater in children with the early-onset persistent AD phenotype [11]. AD children with specific IgE antibodies (extrinsic AD) by 2–4 years of age are at higher risk of the progression of atopic march to allergic rhinitis and asthma than those without (intrinsic AD) [12]. A Canadian birth cohort study reported that a significantly increased risk of FA, asthma and allergic rhinitis was observed in 1-year-old children with AD and allergic sensitization versus those with neither condition [13]. A defective skin barrier, an AD hallmark, has been suggested as a mechanism of atopic march [8,9].
A recent systematic review and meta-analysis of 7 birth cohort studies evaluated AD prevalence across 3 to 6 time points among patients aged 3 months to 26 years and found no significant difference in AD prevalence before versus after childhood [14]. The presence of AD symptoms varied among individuals. Multiple studies found that individuals with early-onset AD were more likely to have symptoms at older ages [14]. The reason for the similar estimated prevalence across ages can be explained by the combination of 3 categories: active disease in both childhood and early adulthood, intermittent disease clearance periods, and later-onset disease.
Two population-based birth cohort studies reported that only a small proportion (~7%) of children with AD experience the complete manifestations of atopic march [15]. However, other studies reported that individuals with early-onset AD are more likely to be symptomatic until later in life, with approximately 17%–31% of patients who developed AD by 2 years of age had AD at all time points up to 18 years of age [15,16]. Approximately 40% of adults with AD have infancy-onset disease, 30% have chronic symptoms into adulthood (early-onset persistent phenotype), and 10% have intermittent symptoms (early-onset intermittent phenotype) [16]. Therefore, it is important to identify high-risk infants with AD that will persist into childhood and adolescence. In addition, early intervention is required to modify atopic march (see Graphical abstract). However, to date, there have been no intervention studies aimed at modifying the atopic march in infants with moderate to severe AD.
Pathogenesis
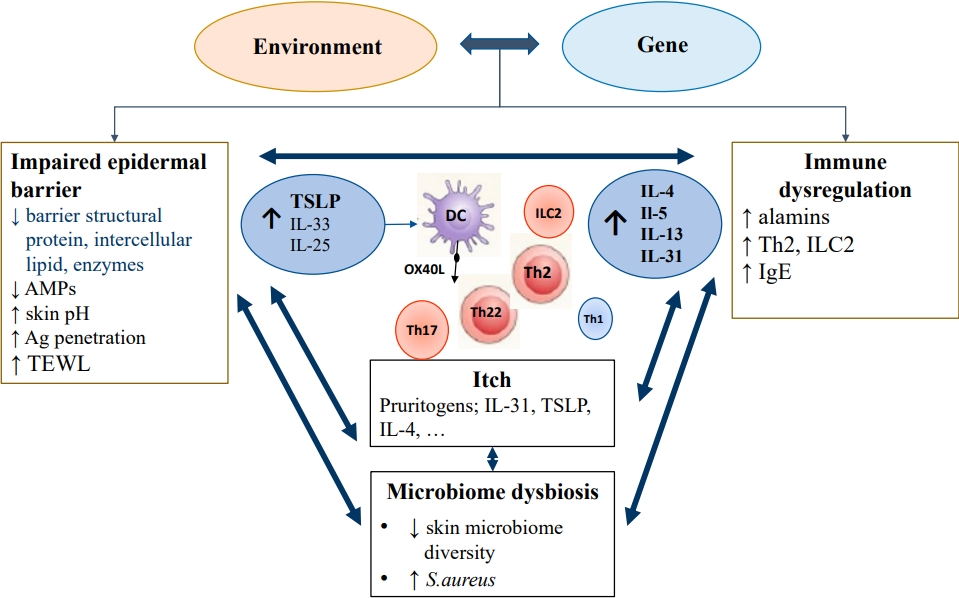
The pathophysiology of AD is complex and multifactorial, caused by interactions between various factors, including epidermal barrier dysfunction, immune dysregulation, microbiome dysbiosis, and pruritus, with strong genetic susceptibility (Fig. 1) [17]. A considerable body of evidence suggests that both epidermal barrier dysfunction and immune deviation to T helper 2 (Th2) inflammation play key roles in AD.
Two hypotheses have been proposed: inside to outside and outside to inside. The first hypothesis is that abnormalities in the innate immune system cause skin inflammation, leading to barrier impairment upon antigen or irritant stimulation. Various mutations and polymorphisms of inflammatory genes have been associated with AD, such as interleukin (IL)-4 receptor α and the cluster of differentiation (CD)-14 genes, the serine protease inhibitor Kazal type 5, regulated on activation, normal T cell expressed and secreted (RANTES), IL-4, and IL-13 [18]. Th2 lymphocyte-dominant immune dysregulation produces IL-4 and IL-13, which inhibit filaggrin (FLG) expression.
The outside to inside hypothesis suggests that an impaired skin barrier is the first step in AD pathogenesis and causes immune dysregulation. FLG is an important structural protein in the stratum corneum (SC) that ensures proper epidermal differentiation and skin barrier function [19]. FLG breakdown products produced in the cornified layer contribute to skin moisture retension, pH regulation, barrier permeability regulation, and microbial protection [19,20]. The FLG loss-of-function mutation and its effects on epidermal integrity provide strong evidence supporting outside to inside hypothesis. While there is ongoing debate regarding the sequence of events, it is evident that both epidermal barrier dysfunction and immune dysregulation significantly contribute to the pathogenesis of AD, as they intricately interact with each other.
Recently, it has been suggested that barrier-initiated AD pathogenesis may induce immune dysregulation, further compromising permeability barrier function and forming a potential vicious outside-inside-outside circle in AD.
Although AD is well known to be characterized by a strong Th2 immune response, it is recently recognized as a more heterogeneous disease with additional involvement of the Th22, Th17/IL-23, and Th1 cytokine pathways depending on disease subtype [21].
The epidermal barrier consists of SC and tight junctions, and SC are composed of corneocytes and the extracellular matrix, called the brick and mortar structure. Intact skin is an important physical and immunological barrier to allergens, microbes, and chemicals. Skin barrier impairment, caused by inherited defects or acquired insults, is characterized by downregulated epidermal barrier structural proteins (including FLG, keratins, loricrin, involucrin, and cell adhesion molecules), decreased intercellular lipids and enzymes, decreased antimicrobial peptides (AMPs), increased skin pH, and reduced skin microbiome diversity with a greater abundance of Staphylococcus aureus [21]. Most patients with AD have reduced epidermal terminal differentiation and SC ceramide levels, either primarily or secondarily by immunemediated mechanisms. A disrupted epithelium exposed to stimuli such as proteolytic allergens, bacteria, parasites, and chemicals promotes antigen penetration and triggers a variety of proteinase-activated receptors and pattern recognition receptors on barrier epithelial cells, inducing the release of epithelial-derived cytokines (alarmins) such as thymic stromal lymphopoietin (TSLP), IL-33, and IL-25. In the epithelial regulation of allergic-type 2 responses, 3 epithelial-derived cytokines are critical mediators of type 2 inflammation through the activation of dendritic cells (DCs) and type 2 innate lymphoid cells (ILC2s) [19-23]. DCs at barrier surfaces present processed allergens to naive T cells in the draining lymph nodes through major histocompatibility complex class II molecules. In the presence of IL-4, naive T cells differentiate into Th2 cells, the major cell type that skews the immune reaction to allergens by producing the cytokines IL-4, IL-5, IL-9, IL-13, and IL-31. Activated Th2 cells and ILC2s release IL-4 and IL-13, which promote IgE class switching [19-23]. In addition, IL-4 and IL-13 stimulate keratinocytes to produce TSLP. TSLP overexpression has been identified in the keratinocytes in both acute and chronic lesions of AD [24]. TSLP activates OX40 ligand-expressing dermal DCs to induce differentiation of naive T cells to inflammatory Th2 cells. The epidermal production of TSLP is correlated with clinically observed lesions and AD severity and persistence [24].
Cytokines and chemokines, such as IL-4, IL-5, IL-13, eotaxins, CC chemokine ligand (CCL)17, CCL18, and CCL22, are produced by Th2 cells and DCs and stimulate the infiltration of DCs, mast cells, and eosinophils into the skin. ILC2s are potent sources of IL-5 and IL-13.
IL-22, an α-helical cytokine belonging to the IL-20 subfamily that is strongly upregulated in AD, is produced by the Th22 cell subset. IL-22 signals via a heterodimer of IL-22 receptor 1 and IL-10 receptor 2, which are expressed on epithelial cells in the skin (keratinocytes), lung, and gut [25]. Increased IL-22 levels act as proinflammatory cytokines, leading to upregulation of AMPs in synergy with IL-17. IL-22 has also been suggested to induce epidermal hyperplasia by promoting keratinocyte proliferation and barrier defects by inhibiting terminal differentiation [26]. IL-22 plays important pathogenic roles in AD initiation and development and is correlated with AD severity.
Th1 and Th17 cells are suggested to play a role, especially in certain subtypes such as intrinsic, pediatric, and Asian phenotypes [27]. However, Th2 and Th22 cells play predominant roles in all AD subtypes [28]. Therefore, dupilumab, which blocks IL-4/IL-13 receptors, is equally effective for extrinsic and intrinsic AD as well as in pediatric and adult AD. Similar or higher Th2 and Th1 activity but much greater Th22 and Th17 immune responses are seen in the lesional skin of patients with intrinsic versus extrinsic AD [29].
Pruritus is the most burdensome symptom in AD, leading to unremitting scratching and further damage to the epithelial barrier, and impairing quality of life of patients and their family. It is primarily a sensory perception of the skin mediated by unmyelinated C-fibers and thinly myelinated Aδ fibers originating from cell bodies in the dorsal root ganglion [30]. It has been suggested that endogenous and exogenous pruritogens such as histamine, 5-hydroxytryptamine, proteases, substance P, various cytokines including IL-31 and TSLP, and environmental allergens can signal through specific itch pathways on nerve fiber endings [24,31]. IL-31 is a potent pruritogenic cytokine in AD. Physical damage due to chronic scratching significantly increases cutaneous TSLP levels. TSLP directly causes pruritus as a pruritogen and indirectly by inducing Th2-related cytokines that activate sensory neurons. Moreover, IL-4 enhances neuronal responsiveness to multiple pruritogens. Therefore, pruritogens, including TSLP and Th2 cytokines, are implicated in AD development and aggravation by inducing itching, scratching, and skin barrier dysfunction [24].
Evidence is growing for an important role of the microbiome in AD pathogenesis: specifically, the abundance of S. aureus and relative reduction of commensal organisms that may play a role in regulating growth of S. aureus [32].
Subtypes
AD is a heterogeneous disease with several phenotypes and endotypes characterized by the activation of diverse cytokine signaling pathways, including Th1, Th2, Th22, and Th17 cells, depending on disease subtype. Phenotypes can be classified according to clinical features, such as age, severity, race, and therapeutic response. An endotype is a subtype of a health condition defined by distinct functional or pathobiological mechanisms such as extrinsic/intrinsic AD based on atopic status. The term endophenotype is used to connect the clinical phenotype and mechanical endotype. Defining a distinct endophenotype is a key to determining personalized therapy. Personalized targeted therapy is possible if there is a unique cytokine signature that characterizes an individual’s endotype. Studies on endophenotype based on race/ethnicity and age have been conducted. However, additional studies with a greater number of subjects are required to elucidate the characteristics of these subtypes. Here, we describe the subtypes according to age at onset, atopic status, and disease chronicity, which have shown several distinct characteristics.
1. Subtypes based on age at onset
The clinical AD phenotypes according to age at onset can be clearly defined. Generally, 4 types are classified: infantile (<2 years), early childhood (2–6 years), late childhood (6–12 years), and adolescence (12–18 years) [33,34].
A European birth cohort study revealed that the prevalence of asthma and FA by 6 years of age strongly increased among children with early phenotypes (aged <2 years), especially those with persistent symptoms [35]. Similarly, a recent Korean study of school-aged children and adolescents with AD found that comorbid FA, allergic rhinitis, and asthma as well as inhalant allergen sensitization were more prevalent in infancy-onset (<2 years of age) than childhood-onset AD (≥2 years of age) [34]. While a significant proportion of patients with the early-onset phenotype can reach complete remission before 2 years of age, another proportion, estimated at up to 40%, continues to suffer from the disease over a long period of time [11], and this category of patients may be at high risk for atopic march [36].
As the immune system changes with age, AD in different age groups may present diverse phenotypes and endotypes. Unique cytokine signatures characterizing individual pediatric endotypes may enable age-specific tailored treatment.
The shape and distribution of lesions in AD vary among age groups: cheeks, scalp, and trunk in infants; extensors of limbs in younger children; flexural distribution of limbs in older children; and additional lichenified lesions on the forehead and neck in adolescents. These changes may be derived from background endotype skewing over time. Therefore, it is crucial to elucidate them to ensure proper treatment.
In addition, even among children of a similar age, the underlying immunological profiles may differ according to atopic status (intrinsic vs. extrinsic), disease duration (acute vs. chronic), severity (mild vs. moderate to severe), and race. However, unlike in adults, making the distinction between extrinsic and intrinsic AD may not be clear in infants and young children because some intrinsic AD cases evolve to the extrinsic type through sensitization.
Studies with peripheral blood samples suggested that infants present overexpression of regulatory T cells and a greater Th17 lineage capacity than adults [37,38]. At birth, immune responses are Th2 polarized, with low Th1/ interferon (IFN)-γ levels in healthy newborns and those with AD. The number of cutaneous lymphocyte antigen (CLA)+ Th1 cells was lower in infants and increased with age. Children (<5 years old) with moderate to severe AD showed suppressed and delayed development of skin-homing (CLA+) Th1 cells in the peripheral blood. CLA+ Th1 cell counts in AD infants were lower than those of age-matched controls and older children with AD [39]. However, frequencies of CLA+ Th2 cells were similarly expanded across all age groups of infants, children, adolescents, and adults with AD and significantly higher than in age-matched controls [40]. After infancy, systemic Th2 cells (CLA-Th2 cells) increased in AD patients of all ages, suggesting systemic immune activation with disease chronicity [40]. In addition, IL-22 frequencies also increased from normal levels in infants to significantly higher levels in adolescents and adults versus their respective control subjects. Principal component analysis of the flow cytometric marker frequencies (percentages) in patients with AD by age showed 3 meaningful age clusters: infants (0–5 years), children and adolescents (6–17 years), and adults (≥18 years), suggesting unique molecular profiles of AD by age [40].
Epidermal hyperplasia is more common in the lesional skin of children younger than 5 years of age who developed AD within 6 months of age than in adults. In addition, the nonlesional skin of infants and young children shows significant hyperplasia compared to that of adults [41]. Epidermal TSLP expression as early as 2 months of age is associated with AD later in life [42]. Taken together, true AD can be initiated before lesional skin appears in children with early-onset AD.
A study of skin samples taken from moderate to severe AD patients of different ages (0–5, 6–11, 12–17, and ≥18 years) found common features of Th2 (Th2-related markers of IL-13, CCL17/thymus and activation-regulated chemokine, CCL18/pulmonary and activation-regulated chemokine, and IL-4R) and Th22 skewing (Th22-related markers of IL22 and S100As) [43]. The differences in expression levels of cytokines between age groups of AD were as follows: infants showed the greatest Th17-related cytokines, whereas long-standing adults displayed Th1 skewing cytokines, including IFN-γ and C-X-C motif chemokine ligand (CXCL)9/CXCL10/ CXCL11, suggesting disease chronicity. The expression level of Th17-related genes was inversely related to the developmental age of children aged 0–11 years with or without AD, was generally higher in the skin of AD patients versus healthy controls, and presented 2 peaks, with the highest expression in infants followed by adults [43]. Although the role of Th17 in AD has not yet been clearly elucidated, IL-17 is less important in AD than in psoriasis. Table 1 shows the characteristics of subtypes by age at onset.
2. Subtypes by atopic status
AD has long been subdivided into extrinsic/atopic and intrinsic/nonatopic subtypes. The extrinsic phenotype (60%–80% of cases) is characterized by high serum total and specific IgE levels, eosinophilia, personal and familial atopic backgrounds, and a higher FLG mutation rate. In contrast, patients with intrinsic AD (20%–40%) have normal IgE levels, no other atopic background, a female predominance, a delayed disease onset, and preserved barrier function [44,45]. However, even in the same extrinsic subtype, there may be differences in the sensitized allergens as well as stage and site of AD lesion according to age. Sensitization to food allergens is common in infants and young children, whereas sensitization to inhaled allergens is more frequent in older children.
Most studies in infants and young children have attempted to characterize disease phenotypes using peripheral blood analysis. The eosinophil count, eosinophil cationic protein level, and IL-5 detection rate were higher in infants with extrinsic versus intrinsic AD [46].
Increased Th1 signals (IFN-γ, CXCL9, CXCL10, and MX-1) and more pronounced Th17/Th22 activation (IL-17A, CCL20, Elafin, and IL-22), which are linked to psoriasis, are noted in intrinsic AD. Levels of antimicrobial activity (S100A9 and S100A12), which are coregulated by IL-17/IL-22, are higher in intrinsic versus extrinsic lesions [47].
In the skin, Th1/IFN-γ-related gene expression and levels of the Th17 chemokine CCL20 are correlated with disease severity in intrinsic AD. On the other hand, Th2 markers were positively correlated with disease severity but negatively correlated with the barrier products of loricrin, periplakin, and FLG in extrinsic AD [46]. Intrinsic AD has inflammatory (IL-22, IL-36α/γ, IL-36RN, and CCL22) and lipid metabolism pathways that overlap with psoriasis, supporting Th17/IL-23 and IL-22 as common profiles of both conditions [47].
Although each type has characteristic cytokine profiles, they share a similar clinical presentation in the lesional skin and a similar increase in Th2 markers; increased infiltration of T cells and DCs in the lesional and nonlesional skin of both AD (more cellular infiltrates of T cells, myeloid DCs, and Langerhans cells in intrinsic AD) and epidermal hyperplasia in the lesional skin versus nonlesional skin [47]. Table 2 summarizes the characteristics of subtypes by atopic status.
3. Subtypes by stage
Skin lesions vary widely, but can be classified as acute or chronic. Acute lesions begin with erythematous papules and serous exudates with intense itching and include secondary lesions, such as excoriations and crusted erosions due to scratching. When acute lesions persist, subacute lesions such as erythematous scaly papules and plaques appear. Progressive itching and rashes result in chronic lichenified lesions characterized by prominent skin marks with hyper- or hypopigmentation.
AD usually presents as multiple lesions of different stages at multiple sites. It is common to find overlapping acute and chronic lesions in the same patient. Acute lesions begin with a marked increase in AMP and a lesser increase in IL-17 levels as well as the upregulation of Th2 and Th22 cytokines. Intensification of the Th2 and Th22 cytokine axes with disease chronicity has been demonstrated along with significant increases in Th1 [48,49]. Taken together, acute inflammation in AD is driven by type 2 cytokines, while enhanced Th2 and Th22 as well as additional Th1 responses are involved in the chronic stage; changes from acute to chronic AD are quantitative rather than qualitative in terms of Th2, Th22, Th1, and Th17 responses, and additional features develop only in chronic inflammation (Table 3) [48,49].
Treatment
Most patients with mild to moderate AD respond to standard topical anti-inflammatory therapies with optimized skincare. Nevertheless, it is often inadequately controlled by the avoidance of irritants or triggers (food and environment), application of emollients, and intensive topical treatments. The importance of treating children with AD comes from the fact that up to 80% of cases begin in infancy or early childhood and AD is an early presentation of the allergic march. Therefore, it is ideal to shift the treatment goal from symptom resolution to modulation of the immune response behind it. Therefore, early systemic treatment requires in young children with immune dysregulation. However, treatment options for this age group are limited due to the lack of the clinical trial data on the effectiveness and long-term safety of new agents. In addition, no clinical studies have determined whether early systemic treatment of immune dysregulation can modify the disease course in infants and young children. Here, we present a descriptive review of currently accepted new systemic therapies, Th2 cytokine receptor antagonists and Janus kinase inhibitors (JAKi), for children with moderate to severe AD. The results of new systemic agents other than those approved in pediatric AD are not discussed here.
1. Th2 cytokine receptor antagonists
IL-4, IL-13, IL-5, and their respective receptors have been targets of drug development strategies to modulate the Th2 response, a core pathway in AD [50]. IL-4 and IL-13 receptors are expressed in neurons and believed to play additional roles in the itch-scratch mechanism [51].
1) IL-4/IL-13 antagonists
Dupilumab is a human monoclonal antibody that inhibits downstream signaling of IL-4 and IL-13 by binding to IL4Rα [50]. IL-4 and IL-13 share a heterodimeric receptor composed of IL-4Rα and IL-13Rα1, known as the type 2 receptor of IL-4 [52]. It is approved (2022, U.S. Food and Drug Administration; 2022, Korean Ministry of Food and Drug Safety) for children aged 6 months and older with moderate to severe AD. Phase 3 studies evaluated the efficacy and safety of dupilumab for the treatment of moderate to severe AD in children and adolescents aged 6 months to 17 years and reported improvement in AD signs and symptoms, including itching, sleep loss, and quality of life (Table 4, Figs. 2 and 3) [53-55]. Adolescents were administered dupilumab (200/300 mg every 2 weeks or 300 mg every 4 weeks) for 16 weeks. The proportion of patients with 75% improvement in the eczema area and severity index score (EASI 75) from baseline was 41.5%, 38.1%, and 8.2% on 2-week and 4-week dupilumab and placebo regimens, respectively (Table 4, Fig. 2) [53]. In a phase 2a open-label sequential cohort study with a phase 3 open-label extension, adolescents with moderate to severe AD were administered dupilumab 2 mg/kg or 4 mg/kg on a weekly basis. The percent change in EASI from baseline to week 52 for the 2 mg/kg and 4 mg/kg regimens was -85% and -84%, respectively [56]. Almost all children reported at least one mild to moderate treatment-emergent adverse event (TEAE) with a dose-related trend; none led to interruption of treatment.
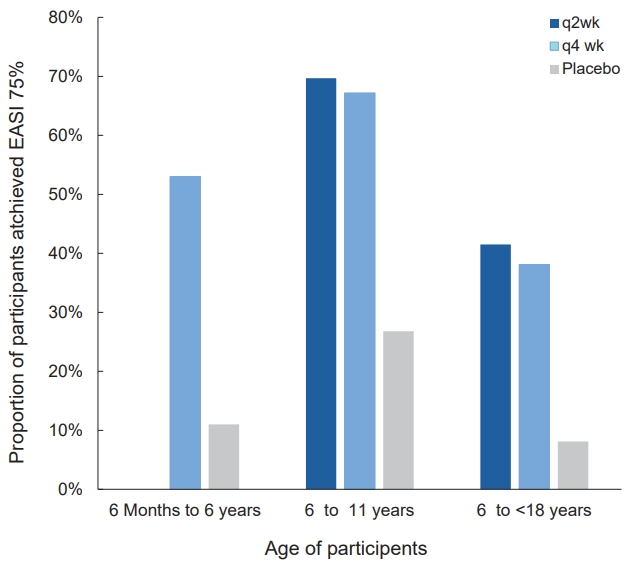
In children aged 6–11 years, the administration of dupilumab with a weight-based regimen of 100/200 mg every 2 weeks (q2 wk) or 300 mg every 4 weeks (q4 wk) for 16 weeks combined with a medium-potency topical corticosteroid (TCS) improved AD; the proportions of patients who achieved an EASI 75 in q2w and q4w dupilumab regimens and placebo were 58.3%, 50.8%, and 12.3%, respectively (Table 4, Fig. 2) [54].
Similar results were reported for younger age groups (6 months to <6 years) with moderate to severe AD. They were given 200/300 mg of dupilumab every 4 weeks for 16 weeks combined with TCS; more patients treated with dupilumab than placebo achieved an EASI 75 (53% vs. 11%) and Investigator’s Global Assessment (IGA) score 0/1 (clear/ almost clear, 28% vs. 4%) (Table 4, Fig. 2) [55]. Itching, one of bothersome symptom, was also remarkably improved in the dupilumab group; the percentage change from baseline in worst itch Numerical Rating Scale score was -49.4% and -2.2% in the dupilumab and placebo groups, respectively. The incidence of conjunctivitis was higher in the dupilumab versus placebo group (5% vs. 0%). However, skin infections, excluding herpes virus infections, were less frequent in the dupilumab versus control group [55].
An ongoing open-label extended phase 3 study (52 weeks) is evaluating the efficacy and long-term safety of dupilumab in patients with moderate to severe AD aged ≥6 months to <18 years participating in 3 parent studies [53,56-58]. Results of dupilumab in adolescents (≥12 to <18 years) among enrolled subjects were recently reported [58]. Patients enrolled in the 3 parent studies and newly enrolled patients received 300 mg of dupilumab every 4 weeks regardless of body weight during the extended study period under the new protocol. At week 52, 42.7% of patients achieved an IGA score of 0/1, while 81% achieved an EASI 75 [58].
Adverse reactions of special interest were mild to moderate and resolved with continued treatment. Clinical trials have reported an increased incidence of conjunctivitis with dupilumab versus placebo. Interestingly, it occurs more frequently in patients with AD versus other diseases such as asthma, chronic rhinosinusitis with nasal polyps, or eosinophilic esophagitis [53,59]. Dupilumab-associated conjunctivitis is less common in children versus adults [60]. However, the exact pathophysiology of conjunctivitis remains unclear.
In summary, dupilumab has a favorable safety profile, even in 6-month-old children and can be administered as a long term therapy in pediatric AD.
2) IL-13 selective antagonist
Selective IL-13 antagonists such as lebrikizumab and tralokinumab can manage AD and improve patients’ quality of life [61]. The 2 agents differ in their binding epitopes and ability to block one or both IL-13 receptors; lebrikizumab does not block 13α2 receptor chains, whereas tralokinumab blocks binding of IL-13 to the IL-13Rα1 and IL-13α2 receptor chains, the decoy receptor, which may be involved in endogenous IL-13 regulation [61].
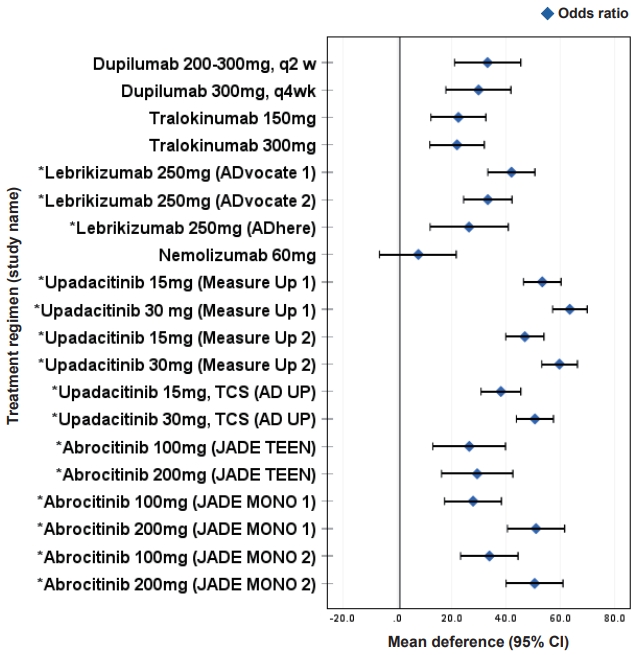
Tralokinumab is a fully humanized antibody targeting IL-13 that blocks its binding to both IL-13Rα1 and IL-13α2 receptor chains [62,63]. It has been approved for the treatment of moderate to severe AD in adults after being studied for up to 52 weeks in phase 3 studies [64,65]. Significant improvements in AD assessment scores, pruritus, sleep interference, and quality of life were noted and maintained over time without the requirement for TCS [64,65]. The results of a phase 3 trial for tralokinumab monotherapy in adolescents (aged 12–17 years) were released [66]. Tralokinumab (150 or 300 mg) was administered every other week; EASI 75 was above 25% by the end of week 16 and reached 44%–64% by the end of maintenance treatment at week 52 with a favorable safety profile. The proportion of patients who achieved an IGA score of 0/1 in the 150 mg and 300 mg tralokinumab and placebo groups was 21.4%, 17.5%, and 4.3%, respectively. The proportion of patients who achieved an EASI 75 in the 150 mg and 300 mg tralokinumab and placebo groups was 28.6%, 27.8%, and 6.4%, respectively (Table 4, Fig. 3). Upper respiratory tract infection was the most common TEAE during the maintenance and safety follow-up period (Table 4) [66].
Lebrikizumab is a fully humanized anti-IL-13 antibody that specifically binds to soluble IL-13 and does not block cytokine binding to the receptor but impairs the heterodimerization of IL-4Rα and IL-13Rα1, thereby inhibiting signal transduction [62,67]. Multicenter double-blind placebo-controlled phase 3 randomized clinical trials evaluated the efficacy of lebrikizumab as monotherapy (ADvocate 1 and 2) and a combination therapy with TCS (Adhere) in the treatment of adolescents (aged ≥12 to <18 years; weight, ≥40 kg) and adults with moderate to severe AD [67,68]. At week 16, EASI 75 in the treatment group vs. placebo was 58.8% vs. 16.2%, 52.1% vs. 18.1% and 69.5% vs. 42.2% in ADvocate1, ADvocate2 and Adhere trials, respectively [67,68]. The TEAEs were mild or moderate in severity and did not lead to discontinuation of the study. The most frequently reposted TEAEs were conjunctivitis (7.4%, 7.5%, 4.9%) and headache (3.2%, 5%, 4.8%) herpes infection (3.2%, 2.8%, 3.4%) in ADvocate1, ADvocate2 and Adhere trials, respectively (Table 4, Fig. 3) [67,68]. A 52-week open label study evaluated the efficacy and safety of lebrikizumab exclusively in adolescent patients of ADvocate1, ADvocate2 and Adhere trials [69]. Serious events leading to treatment discontinuation were infrequent, and 81.9% achieved an EASI 75 by Week 52 [69].
In adults, the improvement in EASI scores after adjusting for placebo was comparable between dupilumab and lebrikizumab (32%–36% and 37%, respectively) and slightly lower in tralokinumab (12%–22%), which may be attributed to differences in the study designs, but because tralokinumab also blocks receptors involved in the endogenous regulation of IL-13 [61,70].
3) IL-31 antagonist
Nemolizumab is a humanized monoclonal antibody against receptor A of IL-31, a prominent pruritogenic cytokine produced by infiltrating Th2 cells in AD, which correlates with disease severity and has been found to be excessively expressed in skin lesions in AD [31,71]. Therefore, IL-31 and its receptor are the focus of strategies to better control the itch-scratch cycle [72,73]. It has been approved for moderate to severe AD over the age of 13 years in adolescents and adults in Japan. In a double-blind, phase 3 trial, 60 mg nemolizumab was subcutaneously administered every 4 weeks with concomitant topical agents to subjects aged 13 years or older with moderate to severe pruritus unresponsive to topical agents, and adolescents aged 13–17 years accounted for approximately 5 percent of subjects [74]. After week 16, the mean percent change in the EASI score was -45.9% with nemolizumab and -33.2% with placebo (Table 4, Fig. 3). Adverse events were generally mild to moderate; however, 1 in the nemolizumab groups discontinued treatment due to AD exacerbation [74]. Although some patients reported exacerbation of AD as an adverse event, these patients also experienced less itching as measured by visual analog scale score [74].
In an open-label phase 2 study of patients aged 12–17 years with moderate to severe AD, nemolizumab was administered subcutaneously as a 60-mg loading dose, followed by 30 mg every 4 weeks until 12 weeks and followed up for 8 more weeks [75]. AD-related proinflammatory biomarkers changed more significantly in EASI responders than in EASI nonresponders [73].
As nemolizumab appears to be a promising agent, large-scale studies are required to evaluate its long-term efficacy and safety. A phase 2 study is ongoing to evaluate the pharmacokinetics, safety, and efficacy of nemolizumab for moderate to severe AD in children aged 2–11 years (NCT04921345).
2. JAK inhibitors
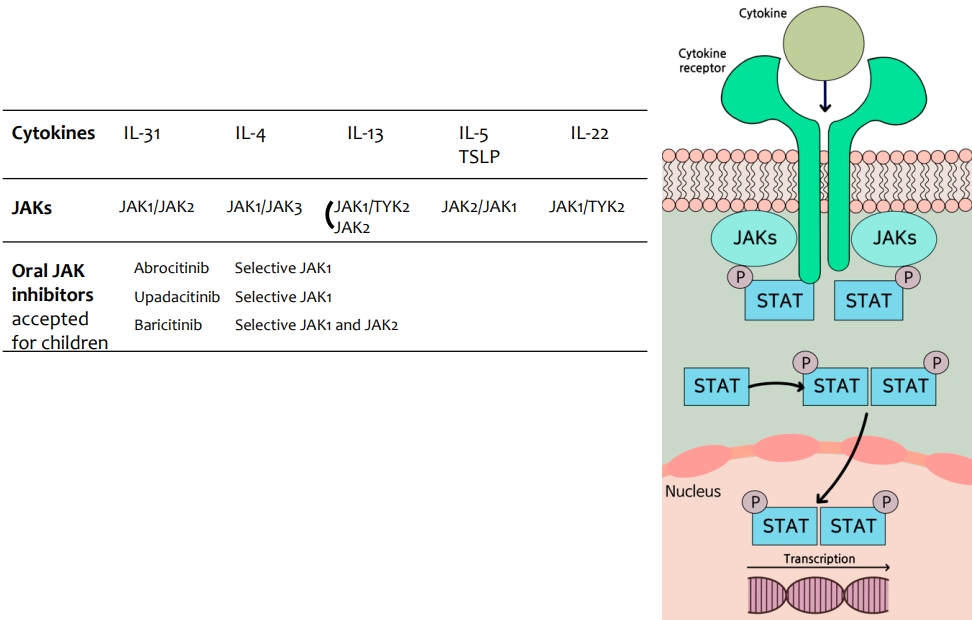
Janus kinase (JAK)s are a group of molecules comprising JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2). Binding of different cytokines to their specific receptor subunits on different cell populations leads to the activation of a specific JAK-signal transducers and activators of transcription (STATs) pathway, and the different isoforms of JAKs are coupled to specific receptor/cytokine pairs. When a cytokine binds to its intracellular domains of type I or type II cytokine receptors, a conformational change is induced, which activates JAK-tyrosine kinases, resulting in phosphorylation of tyrosine residues in the receptor's intracellular domain [40,76]. The phosphorylation of receptor subunits allows for the recruitment of signal molecules, including latent cytoplasmic transcription factors such as STATs, phosphorylated STATs are activated, dimerized, and translocated to the nucleus to regulate target gene expression (Fig. 4). In general, all type I and II receptors rely on JAK1/JAK2 for signal transduction. Depending on the particular receptor, one or more members of the JAK family work together to mediate signal transduction. Therefore, each JAK is often involved in the downstream signaling of multiple cytokine receptors in association with other JAK family members. TYK2 can partner with both JAK1 and JAK2, whereas is a much less widely expressed JAK protein and restricted to receptors containing the common γ chain-containing receptors (Fig. 4).
JAK inhibitors (JAKi) are small molecules that can be administered orally or topically and are recently introduced to treat AD. Currently, more than 90 JAKi are patented, many of which are in clinical development for various indications, such as inflammatory bowel diseases and rheumatoid arthritis [43,76]. As the binding of Th2 and Th22 cytokines to their receptors involves downstream JAK-STAT signaling, JAKi are emerging as attractive compounds for AD treatment.
JAKi approved or under clinical development for AD can be classified into 3 main categories: nonselective (delgocitinib, cerdulatinib, jaktinib, CEE321), dual (baricitinib, ruxolitinib, brepocitinib, ATI-1777), and selective (upadacitinib, abrocitinib, SHR0302) [77].
JAK inhibition exerts a broad immunopharmacological effect by blocking the signal transduction pathways of multiple cytokines. JAKi blocks numerous cytokines that are involved in many aspects of host defense, hematopoiesis, metabolism, cell growth, and cell differentiation; therefore, they can have multiple systemic effects [78]. Serious adverse effects include infections, anemia, pulmonary embolism, malignancy risk, thromboembolic risk, and elevated serum cholesterol [78]. Nonetheless, the hope is a second-generation JAKi with increased selectivity to reduce adverse effects and preserve efficacy. Here, we discuss only orally available JAKi that have been studied in pediatric patients (Table 4, Fig. 3).
Baricitinib is a selective JAK1/JAK2 inhibitor that has been approved for the treatment of patients with moderate to severe AD aged 12 years and older. In 2 independent 16-week phase 3 trials, the participants who achieved EASI 75 reached 24.8% with baricitinib monotherapy and 36.6% with baricitinib plus TCS [79]. The most common adverse events were nasopharyngitis and headache [79]. No cardiovascular disease, venous thromboembolism, or serious hematological changes were detected during the 16-week treatment period. Unlike selective JAK1 inhibitors, no increase in acne incidence was noted [79]. In a pooled safety analysis of cumulative data from 8 adult studies (n= 2,531), simple viral infection and headache were the most frequently reported TEAE, with 2 major cardiovascular events, 2 venous thrombosis events, and 1 death [80]. One death reported after taking baricitinib for more than 12 months was due to gastrointestinal bleeding. The patient was randomized to 1 mg in the first study and then to 4 mg, which was reduced to a renally adjusted dose of 2 mg because of reduced glomerular filtration rate in the long-term extension study. With the satisfactory effect of improvement and onset of action as well as an acceptable safety profile, baricitinib has been studied in children aged 2–17 years with an inadequate response to topical treatment (Table 4) [81]. The baricitinib 4-mg equivalent for 16 weeks achieved a significant improvement in EASI and itching versus placebo.
As hematopoietic signaling of receptors depends crucially on JAK2 homodimers, JAK1 selective inhibitors are suggested as a safer option to avoid major JAKi adverse effects. Upadacitinib and abrocitinib are second-generation JAK1 inhibitors that have been studied in children. Upadacitinib is a selective JAK1 inhibitor approved for the treatment of moderate to severe AD in adults and children aged 12 years. Three phase 3 trials (Measure Up 1, Measure Up 2, and AD Up) evaluated the efficacy of upadacitinib for 16 weeks (15 or 30 mg, once daily) in the treatment of patients aged 12 years or older with moderate to severe AD [82,83]. Measure Up 1 and 2 evaluated upadacitinib as monotherapy, while AD Up examined it with TCS for all participants. The percentage of participants who achieved EASI 75 in Measure Up 1, Measure Up 2, and AD Up was satisfactory with the 15 mg dose (69.6%, 60.1%, and 65 %, respectively), and 30 mg dose (79.7%, 72.9%, and 77%, respectively); both regimens showed statistically significant improvements by week 2. Adverse reactions were mild to moderate in severity and included acne, upper respiratory tract infection, headache, oral herpes, and asymptomatic elevation of plasma creatine phosphokinase. Acne was the most common side effect in all 3 studies. Although acne was not serious and did not lead to treatment discontinuation, it was higher than that observed in previous studies of rheumatoid arthritis, which may be due to the relatively younger age of patients with AD (Table 4) [84,85]. These results demonstrate the potential of upadacitinib as a monotherapy to reduce the burden of TCS use [82,83]. In addition, the incidence of oral herpes infection was lower in upadacitinib monotherapy (3%) than in combination therapy with upadacitinib and TCS [82,83].
Upadacitinib efficacy at week 16 was maintained through the 52-week follow-up studies [86,87]. EASI 75 was achieved in 82.0%, 79.1% and 50.8% of patients maintained on the 15 mg dose and in 84.9%, 84.3% and 69.0% of patients maintained on the 30 mg dose in the Measure Up 1, Measure Up 2 and AD UP studies, respectively. Both doses of upadacitinib were well tolerated, with no new critical safety issues, and a very low rate of treatment discontinuation due to adverse events [86,87]. An open-label multiple dose study in the younger age group (2 to <12 years of age) is currently in progress (NCT 03646604).
Abrocitinib is a JAK1 selective inhibitor that has been approved for the treatment of moderate to severe AD in adolescents and adults. Three phase 3 trials evaluated the efficacy and safety of once-daily 100 mg or 200 mg of abrocitinib for 12 weeks; JADE Mono 1 and 2 studied abrocitinib monotherapy in adolescents and adults (adolescents were 22% and 10% of the study subjects, respectively), whereas JADE TEEN examined abrocitinib plus TCS in adolescents (Table 4, Fig. 3) [88-90].
With the 100-mg dose, the percentage of participants achieved EASI 75 in JADE Mono 1 and 2 as well as JADE TEEN was 40%, 44.5%, and 68.5%, respectively and with 200 mg dose, it was 63%, 61%, and 72%, respectively. Patient-reported signs and symptoms, including sleep loss and quality of life, were substantially improved with abrocitinib monotherapy or combination therapy compared to placebo in adolescents enrolled in JADE TEEN as well as JADE Mono 1 and 2 [90]. Commonly reported adverse events were nausea, vomiting, abdominal pain, headache, increased blood creatine phosphokinase, and acne [88-90]. Thrombocytopenia was noticed in 3 studies and resolved with continued treatment, except for 1 patient who had to withhold treatment for 8 days [88-91]. Regarding the serious side effects of JAKi, no thromboembolism or major cardiovascular events were reported. In upadacitinib studies, the elevation of liver enzymes in the Measure Up 1 and 2 groups and placebo group was 1.7% and 1.1%, respectively, which did not lead to discontinuation of treatment. An integrated safety analysis of a phase 2b study, 4 phase 3 studies and 1 long-term extension study was conducted to evaluate the long-term safety of abrocitinib in adolescents, who represented only 12.7% of all patients in the abrocitinib group [92]. Four events of herpes zoster (0.2%; all in the abrocitinib 200 mg group) resulted in permanent discontinuation of study treatment; abrocitinib 200 mg, age ≥65 years, and severe disease at baseline were associated with higher risk of herpes zoster [92]. Incidence rates presented as numbers of patients with events per 100 patient-years (PYs) were 2.33/100 PY and 2.65/100 PY for serious infection, 4.34/100 PY and 2.04/100 PY for herpes zoster, and 11.83/100 PY and 8.73/100 PY for herpes simplex in the 200-mg and 100-mg groups, respectively. Five venous thromboembolism events occurred (IR 0.30/100 PY) in the 200-mg group [92].
Comparative studies were performed between selective JAKi and dupilumab in adults [93,94]. Upadacitinib 30 mg and abrocitinib 200 mg were superior to dupilumab 300 mg in terms of onset of itch improvement and EASI 75 at 16 weeks. The adverse effects of the 3 drugs were consistent with those reported in previous studies and were mild to moderate in severity. The risk of adverse events was numerically higher in the upadacitinib 30 mg and abrocitinib 200 mg groups than in the dupilumab 300 mg group; however, serious adverse events during treatment were similar across study groups [93,94]. Each drug has its own characteristics including pharmacokinetics. Therefore, this finding needs to be carefully interpreted. JAKi are administered orally every day, and their efficacy is maintained constantly, whereas dupilumab is injected subcutaneously at 4-week intervals; therefore, the concentration before administration is relatively low, and the main outcomes were measured at 4-week intervals, except for weeks 1 and 2.
Conclusion
AD is a heterogeneous systemic inflammatory skin disease associated with immune dysregulation, epithelial barrier dysfunction, and pruritus. Approximately 60%–80% of adults with AD develop the disease during the first 2 years, and 85% develop the disease before 5 years of age, although the rates vary between studies. Up to 40% of patients with infancy-onset AD suffer from the disease into adulthood, and some progress to the atopic march. In addition to concerns about AD chronicity, the systemic Th2 inflammatory response in moderate to severe AD, even at a young age, indicates the need for early appropriate systemic treatment. However, for this very important period in young children, we have limited options for disease intervention. Fortunately, dupilumab, an IL-4 and IL-13 antagonist, has recently been approved for use in children aged 6 months and older. Currently, it is time to focus on whether early treatment for high-risk infants and young children can modify the disease course. For this purpose, background immunologic profiles and clinical features including skin characteristics in young children with AD should be further elucidated.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


