


Introduction
Globally, colorectal cancer (CRC) is the fourth most common cancer in men and the third most common cancer in women1). Major efforts have focused on understanding the underlying pathogenesis of this common cancer. Although most CRC occurs sporadically, genetic predisposition related to its pathogenesis has been reported2). One of the most investigated genotypic subtypes of CRC is the aberrancy of the mismatch repair pathway, usually found in combination with microsatellite instability (MSI)3). Lynch syndrome, also called hereditary nonpolypsosis colorectal cancer (HNPCC), is caused by germline mutation of mismatch repair (MMR) genes4). Individuals affected by Lynch syndrome by heterozygous mutation of MMR usually present CRC in the fourth or fifth decade; however. it is rarely found in teenagers5). K-RAS, a critical oncogene and its product playing a key role in the kinase signaling growth pathway, plays a critical role in the pathogenesis of CRC6).
The revised Bethesda Guidelines recommendations were established for a better understanding and identifying individuals with HNPCC; however, diagnosis of HNPCC at a young age, less than second decades, is difficult as clinician does not suspect it due to its rarity. We recently cared for a female patient with Lynch syndrome, carrying heterozygous MLH1 germline mutation and K-RAS missense somatic mutation; she had CRC at a very early age, 13-years old, and congenital heart disease (transposition of great arteries).
Case report
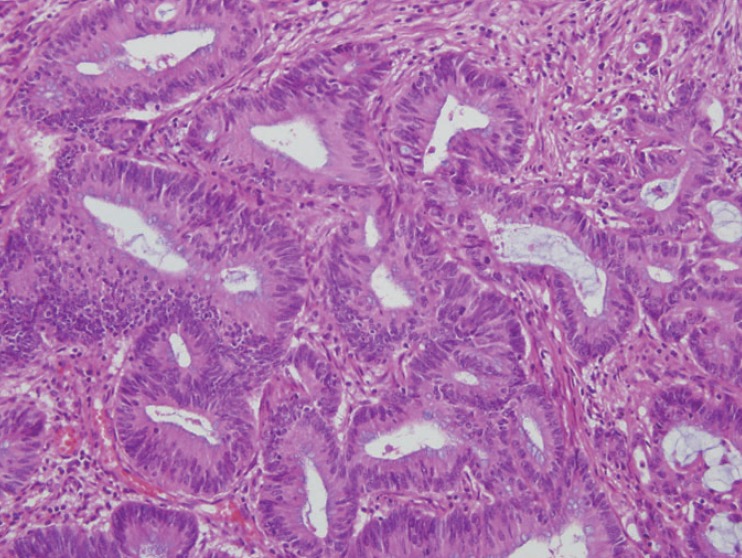
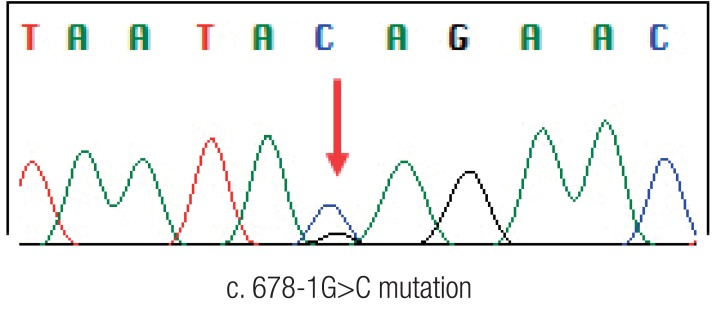
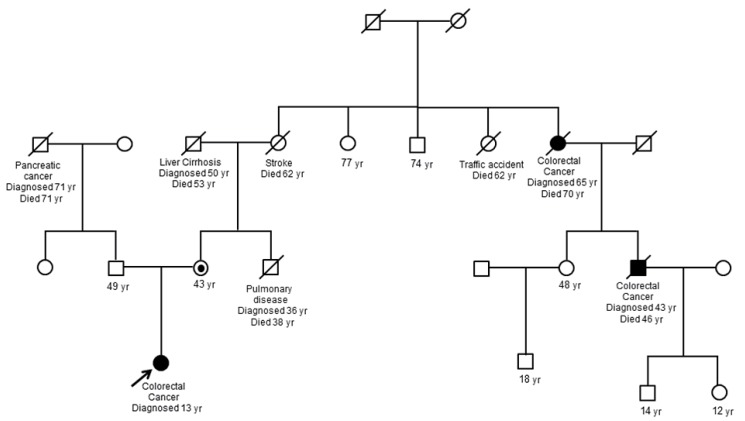
A 13-year-old Korean female was born without complications. Soon after delivery, she was diagnosed with transposition of great arteries and corrective surgery was performed at 3 months of age. She grew up without any specific health problem until she reached 12 years of age. At 13 years of age she visited the hospital for weight loss of 10 kg over 2 months and a pale appearance. She was diagnosed as having Helicobacter pylori gastritis by endoscopy and referred to our hospital for further H. pylori eradication and iron supplementation for management of iron deficiency anemia (hemoglobin, 7.1 g/dL; serum iron, 13 µg/dL; total iron-binding capacity, 381 µg/dL; ferritin, 6.50 ng/dL). She did not have any skin lesions such as café-au-lait spots and physical finding was not remarkable. After treatment, she was free from H. pylori gastritis; however, despite taking a sufficient iron supplement for 1 month, iron deficiency persisted without improvement and palpable mass was noticed. For further evaluation of resistant anemia and palpable abdominal mass, colonoscopy was performed and revealed a huge irregular multilobular mass located on the rectum approximately 6 cm from the anal verge. Biopsy was performed at that site and revealed adenocarcinoma of the rectum. Computed tomography of the abdomen, pelvis, and chest showed a polypoid mass (2.6 cm×2.5 cm) in the rectum and a huge mass (6.7 cm×5.1 cm) in the transverse colon and ascending colon with lymph node enlargement. Total proctocolectomy with ileal pouch anal anastomosis was performed and stage of cancer was T4N2M0. The histopathologic diagnosis was adenocarcinoma of the colon and rectum (Fig. 1). Genetic study of the pathologic specimen revealed a K-RAS gene mutation at codon 12 as Gly12Asp (c. 35G>A) and MSI was found in cancer cells by polymerase chain reaction amplification and fragment analysis by gene analyzer. Germline mutation study of MMR genes using patient's blood revealed a splicing mutation at MLH1 as c. 678-1G>C while other findings were unremarkable (Fig. 2). Genetic study of her parents was performed and revealed that mother had a same MLH1 mutation of patient; however father had none. In addition, 2 cases of CRC were detected on her maternal relatives (Fig. 3).
After informed consent was obtained, 12 cycles of chemotherapy consisting of oxaliplatin (85 mg/m2 on day 1), leucovorin (200 mg/m2 on days 1, 2), and 5-fluorouracil (1,500 mg/m2 on days 1, 2) (FOLFOX) were completed without any specific problems, with the exception of myelosuppression. Diagnostic work-up for recurrence was followed and a suspicious recurrent lesion was noticed at the duodenal second portion and pancreas head portion after completion of the eighth cycle of chemotherapy. After completion of 12 cycles of chemotherapy, imaging study revealed increased mass at the duodenal portion. Supportive management has been provided after chemotherapy and the huge abdominal mass is persistent.
Discussion
Most cancers arise sporadically without apparent causes such as environmental factors or genetic factors. Nonetheless, some cancers arise as the consequence of inherited genetic mutations (germline mutations). Lynch syndrome, caused by germline mutation of MMR genes, such as MLH1, MSH2, MSH6, and PMS2, is an inherited cancer syndrome7,8,9). Lynch syndrome-related tumors, such as CRC and endometrium tumor in females, and a tumor spectrum comprised of hematological malignancies, brain tumors are frequently developed in patients with Lynch syndrome. In these patients, development of CRC by heterozygous mutation of MMR usually occurs during the fourth or fifth decade. However, in recent years, early onset of CRC has been reported in children with either compound heterozygosity or homozygosity for the MMR gene defect10,11,12). In our case CRC by heterozygous germline mutation of MMR gene and a point somatic mutation of the K-RAS developed in teenage without compound heterozygosity or homozygosity for the MMR gene defect. Somatic mutation of K-RAS gene, mainly missense mutation at codons 12 and 13, is found less frequently in CRC with Lynch syndrome than sporadic CRC13). The effect of K-RAS gene mutation on the time of occurrence and prognosis of CRC is uncertain14). However, it could be considered that the mutation on the growth factor signaling pathway (K-RAS) and MMR gene mutation could contribute synergistically to occurrence of CRC at a very early age. Some studies have also shown that age at diagnosis of CRC decreases in successive generations of Lynch families15). This could be applied to the early onset of CRC in this case.
Early diagnosis and management of CRC in teenagers with Lynch syndrome is difficult, since occurrence of CRC is very unusual in this period. In our case, diagnosis was delayed and CRC was managed improperly. The necessity of early screening for detection of CRC in subjects with a family history of early onset CRC, even before two decades, should be emphasized.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link PubMed
PubMed Download Citation
Download Citation


