|
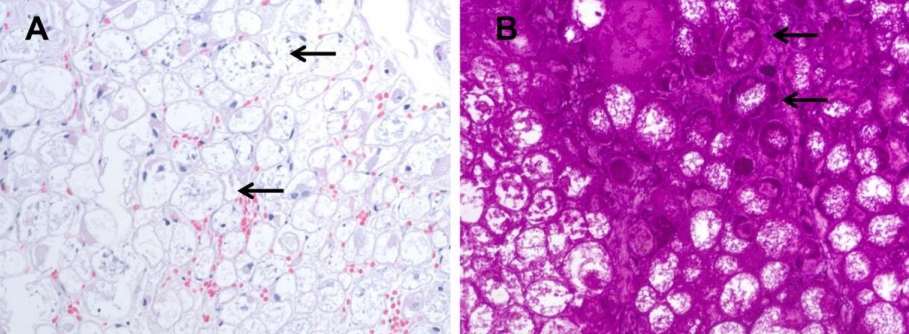
Purpose: Pompe disease (PD) is an autosomal recessive disorder caused by a deficiency of acid alphaglucosidase resulting from pathogenic GAA variants. This study describes the clinical features, genotypes, changes before and after enzyme replacement therapy (ERT), and long-term outcomes in patients with infantile-onset PD (IOPD) and late-onset PD (LOPD) at a tertiary medical center.
Methods: The medical records of 5 Korean... |









")