

Search
- Page Path
-
- HOME
- Search
- Review Article
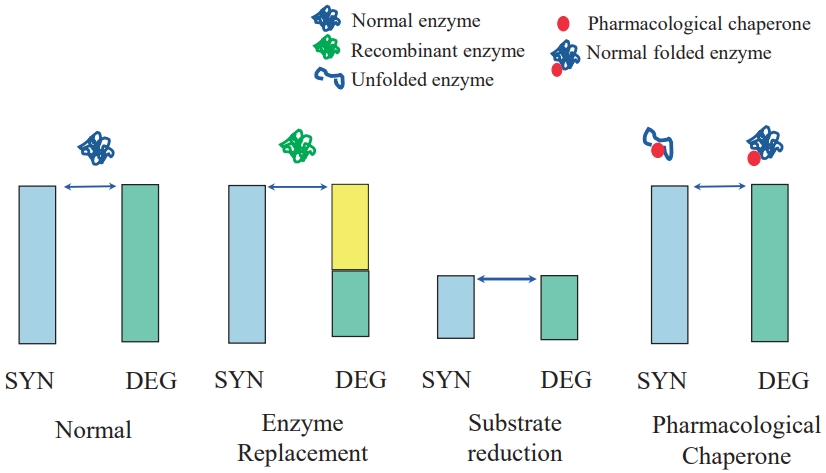
- Original Article
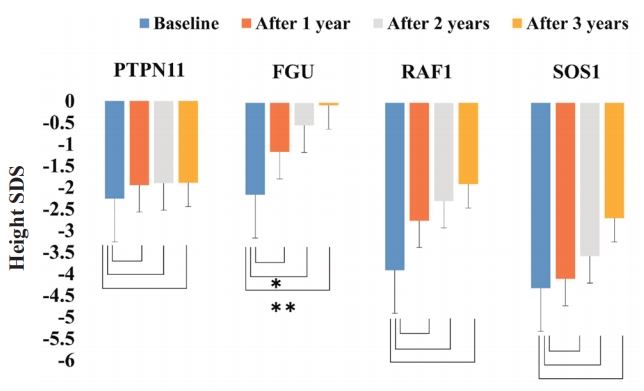
- Review Article
- Case Report
- Original Article
- Case Report
- Original Article
- Case Report
- Original Article
- Case Report
- Original Article
- Case Report
- Original Article
- Review Article
- Case Report
- Original Article
- Case Report
- Original Article
- Case Report
- Original Article
-

-
-
6.02024CiteScore98th percentilePowered by Scopus
-
Impact Factor3.6
-



")


- TOPICS
- ARTICLE CATEGORY
- Editorial Office
-
Korean Pediatric Society
#1606 Seocho World Officetel, 19 Seoun-ro, Seocho-ku, Seoul 06732, Korea
Tel: +82-2-3473-7306 Fax: +82-2-3473-7307 E-mail: office@e-cep.org
Clinical and Experimental Pediatrics is an open access journal. All articles are distributed under the terms of the Creative Commons Attribution NonCommercial License (http://creativecommons.org/licenses/by-nc/4.0/)
Copyright © 2026 by Korean Pediatric Society.

